Analysis of Adaptive Immune Receptor Repertoire Sequencing data with LymphoSeq2
Elena Wu
Shashidhar Ravishankar
David Coffey
2023-06-15
Source:vignettes/LymphoSeq2.Rmd
LymphoSeq2.Rmd
library(LymphoSeq2)
#> Loading required package: data.table
#> Registered S3 methods overwritten by 'ggalt':
#> method from
#> grid.draw.absoluteGrob ggplot2
#> grobHeight.absoluteGrob ggplot2
#> grobWidth.absoluteGrob ggplot2
#> grobX.absoluteGrob ggplot2
#> grobY.absoluteGrob ggplot2
library(RColorBrewer)
library(grDevices)
library(wordcloud2)
library(tidyverse)
#> ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
#> ✔ dplyr 1.1.2 ✔ readr 2.1.4
#> ✔ forcats 1.0.0 ✔ stringr 1.5.0
#> ✔ ggplot2 3.4.2 ✔ tibble 3.2.1
#> ✔ lubridate 1.9.2 ✔ tidyr 1.3.0
#> ✔ purrr 1.0.1
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> ✖ dplyr::between() masks data.table::between()
#> ✖ dplyr::filter() masks stats::filter()
#> ✖ dplyr::first() masks data.table::first()
#> ✖ lubridate::hour() masks data.table::hour()
#> ✖ lubridate::isoweek() masks data.table::isoweek()
#> ✖ dplyr::lag() masks stats::lag()
#> ✖ dplyr::last() masks data.table::last()
#> ✖ lubridate::mday() masks data.table::mday()
#> ✖ lubridate::minute() masks data.table::minute()
#> ✖ lubridate::month() masks data.table::month()
#> ✖ lubridate::quarter() masks data.table::quarter()
#> ✖ lubridate::second() masks data.table::second()
#> ✖ purrr::transpose() masks data.table::transpose()
#> ✖ lubridate::wday() masks data.table::wday()
#> ✖ lubridate::week() masks data.table::week()
#> ✖ lubridate::yday() masks data.table::yday()
#> ✖ lubridate::year() masks data.table::year()
#> ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library(vroom)
#>
#> Attaching package: 'vroom'
#>
#> The following objects are masked from 'package:readr':
#>
#> as.col_spec, col_character, col_date, col_datetime, col_double,
#> col_factor, col_guess, col_integer, col_logical, col_number,
#> col_skip, col_time, cols, cols_condense, cols_only, date_names,
#> date_names_lang, date_names_langs, default_locale, fwf_cols,
#> fwf_empty, fwf_positions, fwf_widths, locale, output_column,
#> problems, specAdaptive Immune Receptor Repertoire Sequencing (AIRR-seq) provides a unique opportunity to interrogate the adaptive immune repertoire under various clinical conditions. The utility offered by this technology has quickly garnered interest from a community of clinicians and researchers investigating the immunological landscapes of a large spectrum of health and disease states. LymphoSeq2 is a toolkit that allows users to import, manipulate and visualize AIRR-Seq data from various AIRR-Seq assays such as Adaptive ImmunoSEQ and BGI-IRSeq, with support for 10X VDJ sequencing coming soon. The platform also supports the importing of AIRR-seq data processed using the MiXCR pipeline. The vignette highlights some of the key features of LymphoSeq2.
Importing data
The function readImmunoSeq imports AIRR-seq receptor
files from Adaptive ImmunoSEQ assay as well well as BGI-IRSeq assay. The
sequences can be (.tsv) files processed using one of the three following
platforms: Adaptive Biotechnologies ImmunoSEQ analyzer, BGI IR-SEQ
iMonitor platform, and the MiXCR pipeline for AIRR-seq data analysis.
The function has the ability to identify file type based on the headers
provided in the (.tsv) file, accordingly the data is transformed into a
format that is compatible AIRR-Community guidelines (https://github.com/airr-community/airr-standards).
To explore the features of LymphoSeq2, this package includes 2 example data sets. The first is a data set of T cell receptor beta (TCRB) sequencing from 10 blood samples acquired serially from a single patient who underwent a bone marrow transplant (Kanakry, C.G., et al. JCI Insight 2016;1(5):pii: e86252). The second, is a data set of B cell receptor immunoglobulin heavy (IGH) chain sequencing from Burkitt lymphoma tumor biopsies acquired from 10 different individuals (Lombardo, K.A., et al. Blood Advances 2017 1:535-544). To improve performance, both data sets contain only the top 1,000 most frequent sequences. The complete data sets are publicly available through Adapatives’ immuneACCESS portal. As shown in the example below, you can specify the path to the example data sets using the command
system.file("extdata", "TCRB_sequencing", package = "LymphoSeq2") #For the TCRB files
#> [1] "/home/runner/work/_temp/Library/LymphoSeq2/extdata/TCRB_sequencing"
system.file("extdata", "IGH_sequencing", package = "LymphoSeq2") #For the IGH files.
#> [1] "/home/runner/work/_temp/Library/LymphoSeq2/extdata/IGH_sequencing"readImmunoSeq can take as input, a single file name, a
list of files or the path to a directory containing AIRR-seq data. The
columns are renamed to follow AIRR-community guidelines based on the
input file type. The function returns a tibble with individual file
names set as the repertoire_id. The CDR3 nucleotide and
amino acid sequences are denoted by the junction and
junction_aa fields respectively. The counts of the CDR3
sequences observed, and their frequency in each individual repertoire is
denoted by the duplicate_count and
duplicate_frequency field respectively.
study_files <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeq2")
study_table <- LymphoSeq2::readImmunoSeq(study_files, threads = 1) %>%
topSeqs(top = 100)Looking at the study_table we see a tibble with 145
columns and 1000 rows
study_table
#> # A tibble: 1,000 × 145
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_CD4_949_4 GAGTCAG… CASSESAGST… FALSE FALSE NA FALSE
#> 2 TRB_CD4_949_5 GCCCTCA… NA FALSE TRUE NA TRUE
#> 3 TRB_CD4_949_6 ATTCCCT… NA FALSE TRUE NA TRUE
#> 4 TRB_CD4_949_7 GTGACAT… CASSPRQGES… FALSE FALSE NA FALSE
#> 5 TRB_CD4_949_8 ACCTTGG… CASSLDGQGQ… FALSE FALSE NA FALSE
#> 6 TRB_CD4_949_9 GTGACCA… CSAKTSGITY… FALSE FALSE NA FALSE
#> 7 TRB_CD4_949_… ACCCTGC… CASSQD*ASS… FALSE TRUE NA TRUE
#> 8 TRB_CD4_949_… CTCCTTC… CAWSDFQGPR… FALSE FALSE NA FALSE
#> 9 TRB_CD4_949_… CTGACGA… CASSPDKWGY… FALSE FALSE NA FALSE
#> 10 TRB_CD4_949_… TCAGAAC… CASSFRTGPT… FALSE FALSE NA FALSE
#> # ℹ 990 more rows
#> # ℹ 138 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …Since the study table is a tibble, we can use tidyverse syntax to extract a list of sample names
Subsetting Data
The tibble structure of the TCR data allows for easy subsampling of
data. To select the TCR sequences from any given samples in the dataset,
the filter function from the dplyr package can
be used.
TRB_Unsorted_0 <- study_table %>%
dplyr::filter(repertoire_id == "TRB_Unsorted_0")
TRB_Unsorted_0
#> # A tibble: 100 × 145
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_Unsorted… TCAATTC… NA FALSE TRUE NA TRUE
#> 2 TRB_Unsorted… CTGATTC… CASSPVSNEQ… FALSE FALSE NA FALSE
#> 3 TRB_Unsorted… ATCAATT… CASSQEVPPY… FALSE FALSE NA FALSE
#> 4 TRB_Unsorted… CACACCC… CASSQEASGR… FALSE FALSE NA FALSE
#> 5 TRB_Unsorted… TGCCATC… NA FALSE TRUE NA TRUE
#> 6 TRB_Unsorted… GCCAGCA… CASSLEHTGA… FALSE FALSE NA FALSE
#> 7 TRB_Unsorted… CCCCTGA… CASSPGDEQYF FALSE FALSE NA FALSE
#> 8 TRB_Unsorted… AGTGCCC… CSARSPSTGT… FALSE FALSE NA FALSE
#> 9 TRB_Unsorted… GGAGCTT… NA FALSE TRUE NA TRUE
#> 10 TRB_Unsorted… CTGTAGT… CASSEKREGH… FALSE FALSE NA FALSE
#> # ℹ 90 more rows
#> # ℹ 138 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …The str_detect function from stringr
package can be used in conjunction with the filter to find
samples using a pattern
CMV <- study_table %>%
dplyr::filter(str_detect(repertoire_id, "CMV"))
CMV
#> # A tibble: 100 × 145
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_CD8_CMV_… CAGCGCA… CASSPPTGER… FALSE FALSE NA FALSE
#> 2 TRB_CD8_CMV_… CAGCCCT… CASSPAGAYY… FALSE FALSE NA FALSE
#> 3 TRB_CD8_CMV_… CAGCCTG… CASSQDWERL… FALSE FALSE NA FALSE
#> 4 TRB_CD8_CMV_… TCGGCCC… CASSQDLMTV… FALSE FALSE NA FALSE
#> 5 TRB_CD8_CMV_… ATCCTGG… CASSLQGREK… FALSE FALSE NA FALSE
#> 6 TRB_CD8_CMV_… GAGGATC… NA FALSE TRUE NA TRUE
#> 7 TRB_CD8_CMV_… ACCCTGC… CASSQDLGQA… FALSE FALSE NA FALSE
#> 8 TRB_CD8_CMV_… GAGTCCG… CASSLAGDSQ… FALSE FALSE NA FALSE
#> 9 TRB_CD8_CMV_… CTCCTCA… CAISDTGELFF FALSE FALSE NA FALSE
#> 10 TRB_CD8_CMV_… TCCAGCC… NA FALSE TRUE NA TRUE
#> # ℹ 90 more rows
#> # ℹ 138 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …A metadata file for the TCR sequencing samples can easily be combined
with the study_table by reading in the metadata file as a
tibble and using the dplyr::left_join function to merge the
two tables. In the example below, a metadata file is imported for the
example TCRB data set which contains information on the number of days
post bone marrow transplant the sample was collected and the cellular
phenotype the blood sample was sorted for prior to sequencing.
TCRB_metadata <- readr::read_csv(system.file("extdata", "TCRB_metadata.csv", package = "LymphoSeq2"), show_col_types = FALSE)
TCRB_metadata
#> # A tibble: 10 × 3
#> samples day phenotype
#> <chr> <dbl> <chr>
#> 1 TRB_Unsorted_0 0 Unsorted
#> 2 TRB_Unsorted_32 32 Unsorted
#> 3 TRB_Unsorted_83 82 Unsorted
#> 4 TRB_CD8_CMV_369 369 CD8+CMV+
#> 5 TRB_Unsorted_369 369 Unsorted
#> 6 TRB_CD4_949 949 CD4+
#> 7 TRB_CD8_949 949 CD8+
#> 8 TRB_Unsorted_949 949 Unsorted
#> 9 TRB_Unsorted_1320 1320 Unsorted
#> 10 TRB_Unsorted_1496 1496 Unsorted
study_table <- dplyr::left_join(study_table, TCRB_metadata, by = c("repertoire_id" = "samples"))
study_table
#> # A tibble: 1,000 × 147
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_CD4_949_4 GAGTCAG… CASSESAGST… FALSE FALSE NA FALSE
#> 2 TRB_CD4_949_5 GCCCTCA… NA FALSE TRUE NA TRUE
#> 3 TRB_CD4_949_6 ATTCCCT… NA FALSE TRUE NA TRUE
#> 4 TRB_CD4_949_7 GTGACAT… CASSPRQGES… FALSE FALSE NA FALSE
#> 5 TRB_CD4_949_8 ACCTTGG… CASSLDGQGQ… FALSE FALSE NA FALSE
#> 6 TRB_CD4_949_9 GTGACCA… CSAKTSGITY… FALSE FALSE NA FALSE
#> 7 TRB_CD4_949_… ACCCTGC… CASSQD*ASS… FALSE TRUE NA TRUE
#> 8 TRB_CD4_949_… CTCCTTC… CAWSDFQGPR… FALSE FALSE NA FALSE
#> 9 TRB_CD4_949_… CTGACGA… CASSPDKWGY… FALSE FALSE NA FALSE
#> 10 TRB_CD4_949_… TCAGAAC… CASSFRTGPT… FALSE FALSE NA FALSE
#> # ℹ 990 more rows
#> # ℹ 140 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …Now the metadata information can be used to further subset the data. For instance to select all “Unsorted” samples collected more than 300 days after bone marrow transplant, we would use the following code
unsorted_300 <- study_table %>%
dplyr::filter(day > 300 & phenotype == "Unsorted")
unsorted_300
#> # A tibble: 400 × 147
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_Unsorted… CAGCCCT… CASSPAGAYY… FALSE FALSE NA FALSE
#> 2 TRB_Unsorted… CAGCGCA… CASSPPTGER… FALSE FALSE NA FALSE
#> 3 TRB_Unsorted… GAGGATC… NA FALSE TRUE NA TRUE
#> 4 TRB_Unsorted… ATCCTGG… CASSLQGREK… FALSE FALSE NA FALSE
#> 5 TRB_Unsorted… GAGTCAG… CASSESAGST… FALSE FALSE NA FALSE
#> 6 TRB_Unsorted… CAGCCTG… CASSQDWERL… FALSE FALSE NA FALSE
#> 7 TRB_Unsorted… GAGTCCG… CASSLAGDSQ… FALSE FALSE NA FALSE
#> 8 TRB_Unsorted… GCCCTCA… NA FALSE TRUE NA TRUE
#> 9 TRB_Unsorted… TCGGCCC… CASSQDLMTV… FALSE FALSE NA FALSE
#> 10 TRB_Unsorted… CTCAGGC… CASSYVGDGY… FALSE FALSE NA FALSE
#> # ℹ 390 more rows
#> # ℹ 140 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …Extracting productive sequences
When AIRR-seq samples are derived from genomic DNA rather than
complimentary DNA made from RNA, then you will find productive and
unproductive sequences. Productive sequences are defined as in-frame
sequences without any early stop codons. To filter out these productive
sequences, you can use the productiveSeq to remove
unproductive sequences and recompute the duplicate_frequency to reflect
the productive amino acid or nucleotide sequence frequencies.
If you are interested in just the complementarity determining region
3 (CDR3) amino acid sequences, then set aggregate to
junction_aa and the duplicate_count for
duplicate amino acid sequences will be summed. The resulting tibble will
have junction_aa, duplicate_count,
duplicate_frequency, reading_frame, and the
most frequent VDJ gene combinations for each of the duplicated amino
acid sequences and the corresponding gene family names. These gene names
are only kept for consistency of the tibble structure, but since a
single amino acid sequence can be generated from different VDJ
combinations, it is inadvisable to use these values for downstream
analysis
aa_table <- LymphoSeq2::productiveSeq(study_table = study_table, aggregate = "junction_aa", prevalence = FALSE)
aa_table
#> # A tibble: 810 × 11
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_CD4_949 CAISVGGSSPLHF TRBV1… TRBD2… TRBJ1… TRBV10 TRBD2 TRBJ1
#> 2 TRB_CD4_949 CASDGGFRNTIYF TRBV1… TRBD2… TRBJ1… TRBV19 TRBD2 TRBJ1
#> 3 TRB_CD4_949 CASGGLNTEAFF NA NA TRBJ1… NA NA TRBJ1
#> 4 TRB_CD4_949 CASGLVAGSTLGGE… TRBV1… TRBD2… TRBJ2… TRBV12 TRBD2 TRBJ2
#> 5 TRB_CD4_949 CASGTGGETQYF TRBV6… TRBD2… TRBJ2… TRBV6 TRBD2 TRBJ2
#> 6 TRB_CD4_949 CASHSSGNTIYF TRBV6… NA TRBJ1… TRBV6 NA TRBJ1
#> 7 TRB_CD4_949 CASKPPGQGGYGYTF TRBV6… TRBD1… TRBJ1… TRBV6 TRBD1 TRBJ1
#> 8 TRB_CD4_949 CASMIDPSGNTIYF TRBV5… NA TRBJ1… TRBV5 NA TRBJ1
#> 9 TRB_CD4_949 CASNARVDSPLHF TRBV6… TRBD1… TRBJ1… TRBV6 TRBD1 TRBJ1
#> 10 TRB_CD4_949 CASRLGESPLHF NA NA TRBJ1… NA NA TRBJ1
#> # ℹ 800 more rows
#> # ℹ 3 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>Alternatively you can aggregate by junction to group
sequences by CDR3 nucleotide sequences. This option will produce a
tibble similar to the output of readImmunoSeq. Many of the
functions within LymphoSeq2 use the results from
productiveSeq function. Please be sure to check the
function documentation.
nuc_table <- LymphoSeq2::productiveSeq(study_table = study_table, aggregate = "junction",
prevalence = FALSE)
nuc_table
#> # A tibble: 812 × 12
#> repertoire_id junction junction_aa v_call d_call j_call v_family d_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_CD4_949 AACCTGAGCTC… CASSVEVGSA… TRBV9… NA TRBJ1… TRBV9 NA
#> 2 TRB_CD4_949 AACCTGAGCTC… CASSVMVGTE… TRBV9… NA TRBJ1… TRBV9 NA
#> 3 TRB_CD4_949 AACGCCTTGGA… CASSDSGVPG… TRBV5… TRBD1… TRBJ1… TRBV5 TRBD1
#> 4 TRB_CD4_949 AACGCCTTGTT… CASSSQGLNT… TRBV5… TRBD1… TRBJ2… TRBV5 TRBD1
#> 5 TRB_CD4_949 AACGCCTTGTT… CASSLTGRSD… TRBV5… NA TRBJ2… TRBV5 NA
#> 6 TRB_CD4_949 AAGATCCAGCC… CASSSNPDQP… NA NA TRBJ1… NA NA
#> 7 TRB_CD4_949 AATCTTCACAT… CASSQGGPLHF NA TRBD2… TRBJ1… NA TRBD2
#> 8 TRB_CD4_949 AATGTGAACGC… CASSLAGNTE… TRBV5… TRBD2… TRBJ1… TRBV5 TRBD2
#> 9 TRB_CD4_949 AATTCCCTGGA… CASSQPGLTN… NA TRBD1… TRBJ1… NA TRBD1
#> 10 TRB_CD4_949 AATTCCCTGGA… CASSQGGSYN… NA NA TRBJ1… NA NA
#> # ℹ 802 more rows
#> # ℹ 4 more variables: j_family <chr>, reading_frame <chr>,
#> # duplicate_count <dbl>, duplicate_frequency <dbl>If the parameter prevalence is set to TRUE, then a new column is added to each of the data frames giving the prevalence of each TCR beta CDR3 amino acid sequence in 55 healthy donor peripheral blood samples. Values range from 0 to 100 percent where 100 percent means the sequence appeared in the blood of all 55 individuals.
Notice in the example below that there are no amino acid sequences given in the first and fourth row of the study_table table for sample “TRB_Unsorted_949”. This is because the nucleotide sequence is out of frame and does not produce a productively transcribed amino acid sequence. If an asterisk (*) appears in the amino acid sequences, this would indicate an early stop codon.
study_table %>%
dplyr::filter(repertoire_id == "TRB_Unsorted_0")
#> # A tibble: 100 × 147
#> sequence_id sequence sequence_aa rev_comp productive vj_in_frame stop_codon
#> <chr> <chr> <chr> <lgl> <lgl> <lgl> <lgl>
#> 1 TRB_Unsorted… TCAATTC… NA FALSE TRUE NA TRUE
#> 2 TRB_Unsorted… CTGATTC… CASSPVSNEQ… FALSE FALSE NA FALSE
#> 3 TRB_Unsorted… ATCAATT… CASSQEVPPY… FALSE FALSE NA FALSE
#> 4 TRB_Unsorted… CACACCC… CASSQEASGR… FALSE FALSE NA FALSE
#> 5 TRB_Unsorted… TGCCATC… NA FALSE TRUE NA TRUE
#> 6 TRB_Unsorted… GCCAGCA… CASSLEHTGA… FALSE FALSE NA FALSE
#> 7 TRB_Unsorted… CCCCTGA… CASSPGDEQYF FALSE FALSE NA FALSE
#> 8 TRB_Unsorted… AGTGCCC… CSARSPSTGT… FALSE FALSE NA FALSE
#> 9 TRB_Unsorted… GGAGCTT… NA FALSE TRUE NA TRUE
#> 10 TRB_Unsorted… CTGTAGT… CASSEKREGH… FALSE FALSE NA FALSE
#> # ℹ 90 more rows
#> # ℹ 140 more variables: complete_vdj <lgl>, locus <chr>, v_call <chr>,
#> # d_call <chr>, d2_call <chr>, j_call <chr>, c_call <chr>,
#> # sequence_alignment <chr>, sequence_alignment_aa <chr>,
#> # germline_alignment <chr>, germline_alignment_aa <chr>, junction <chr>,
#> # junction_aa <chr>, np1 <chr>, np1_aa <chr>, np2 <chr>, np2_aa <chr>,
#> # np3 <chr>, np3_aa <chr>, cdr1 <chr>, cdr1_aa <chr>, cdr2 <chr>, …After productiveSeq is run, the unproductive sequences
are removed and the duplicate_frequency is recalculated for
each sequence. If there were two identical amino acid sequences that
differed in their nucleotide sequence, they would be combined and their
counts added together.
aa_table %>%
dplyr::filter(repertoire_id == "TRB_Unsorted_0")
#> # A tibble: 83 × 11
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_Unsorted_0 CAISDLAVPPSYN… TRBV1… TRBD2… TRBJ2… TRBV10 TRBD2 TRBJ2
#> 2 TRB_Unsorted_0 CARPPYWDYGYTF TRBV1… NA TRBJ1… TRBV10 NA TRBJ1
#> 3 TRB_Unsorted_0 CASKYGGAEKLFF TRBV7… TRBD2… TRBJ1… TRBV7 TRBD2 TRBJ1
#> 4 TRB_Unsorted_0 CASREAWTATNEK… TRBV2… TRBD1… TRBJ1… TRBV2 TRBD1 TRBJ1
#> 5 TRB_Unsorted_0 CASRHREANYGYTF TRBV2… NA TRBJ1… TRBV28 NA TRBJ1
#> 6 TRB_Unsorted_0 CASRPDRGSSPLHF TRBV2… TRBD1… TRBJ1… TRBV28 TRBD1 TRBJ1
#> 7 TRB_Unsorted_0 CASRPGQGVGEQYF TRBV1… TRBD1… TRBJ2… TRBV10 TRBD1 TRBJ2
#> 8 TRB_Unsorted_0 CASRPTKNSDGEL… TRBV1… NA TRBJ2… TRBV19 NA TRBJ2
#> 9 TRB_Unsorted_0 CASRSGRTNQPQHF TRBV2… TRBD2… TRBJ1… TRBV2 TRBD2 TRBJ1
#> 10 TRB_Unsorted_0 CASSARSYEQYF TRBV7… NA TRBJ2… TRBV7 NA TRBJ2
#> # ℹ 73 more rows
#> # ℹ 3 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>Create a table of summary statistics
To create a table summarizing the total number of sequences, number
of unique productive sequences, number of genomes, clonality, Gini
coefficient, and the frequency (%) of the top productive sequence,
Simpson index, Inverse Simpson index, Hill diversity index, Chao1 index
and Kemp index in each imported file, use the function
clonality.
LymphoSeq2::clonality(study_table = study_table)
#> # A tibble: 10 × 8
#> repertoire_id total_sequences unique_productive_se…¹ total_count clonality
#> <chr> <int> <int> <dbl> <dbl>
#> 1 TRB_CD4_949 100 80 23093 0.349
#> 2 TRB_CD8_949 100 81 23072 0.292
#> 3 TRB_CD8_CMV_369 100 78 1456 0.305
#> 4 TRB_Unsorted_0 100 83 14776 0.128
#> 5 TRB_Unsorted_13… 100 83 157660 0.279
#> 6 TRB_Unsorted_14… 100 82 28876 0.260
#> 7 TRB_Unsorted_32 100 82 17043 0.105
#> 8 TRB_Unsorted_369 100 80 274812 0.387
#> 9 TRB_Unsorted_83 100 81 170526 0.328
#> 10 TRB_Unsorted_949 100 82 4971 0.247
#> # ℹ abbreviated name: ¹unique_productive_sequences
#> # ℹ 3 more variables: gini_coefficient <dbl>, top_productive_sequence <dbl>,
#> # convergence <dbl>The clonality score is derived from the Shannon entropy, which is calculated from the frequencies of all productive sequences divided by the logarithm of the total number of unique productive sequences. This normalized entropy value is then inverted (1 - normalized entropy) to produce the clonality metric.
The Gini coefficient, Chao1 estimate, Kemp estimate, Hill estimate, Simpson index and Inverse Simpson index are alternative metric to measure sequence diversity within the immune repertoire.
The Gini coefficient is an alternative metric used to calculate repertoire diversity and is derived from the Lorenz curve. The Lorenz curve is drawn such that x-axis represents the cumulative percentage of unique sequences and the y-axis represents the cumulative percentage of reads. A line passing through the origin with a slope of 1 reflects equal frequencies of all clones. The Gini coefficient is the ratio of the area between the line of equality and the observed Lorenz curve over the total area under the line of equality.
Calculate clonal relatedness
One of the drawbacks of the clonality metric is that it does not take into account sequence similarity. This is particularly important when studying affinity maturation or B cell malignancies(Lombardo, K.A., et al. Blood Advances 2017 1:535-544). Clonal relatedness is a useful metric that takes into account sequence similarity without regard for clonal frequency. It is defined as the proportion of nucleotide sequences that are related by a defined edit distance threshold. The value ranges from 0 to 1 where 0 indicates no sequences are related and 1 indicates all sequences are related. Edit distance is a way of quantifying how dissimilar two sequences are to one another by counting the minimum number of operations required to transform one sequence into the other. For example, an edit distance of 0 means the sequences are identical and an edit distance of 1 indicates that the sequences different by a single amino acid or nucleotide.
IGH_path <- system.file("extdata", "IGH_sequencing", package = "LymphoSeq2")
IGH_table <- LymphoSeq2::readImmunoSeq(path = IGH_path, threads = 1) %>%
LymphoSeq2::topSeqs(top = 100)
LymphoSeq2::clonalRelatedness(study_table = IGH_table, edit_distance = 10)
#> # A tibble: 10 × 2
#> repertoire_id relatedness
#> <chr> <dbl>
#> 1 IGH_MVQ108911A_BL 0.61
#> 2 IGH_MVQ194745A_BL 0.7
#> 3 IGH_MVQ81231A_BL 0.61
#> 4 IGH_MVQ89037A_BL 0.35
#> 5 IGH_MVQ90143A_BL 0.03
#> 6 IGH_MVQ92552A_BL 0.08
#> 7 IGH_MVQ93505A_BL 0.31
#> 8 IGH_MVQ93631A_BL 0.83
#> 9 IGH_MVQ94865A_BL 0.06
#> 10 IGH_MVQ95413A_BL 0.01Draw a phylogenetic tree
A phylogenetic tree is a useful way to visualize the similarity
between sequences. The phyloTree function create a
phylogenetic tree of a single sample using neighbor joining tree
estimation for amino acid or nucleotide CDR3 sequences. Each leaf in the
tree represents a sequence color coded by the V, D, and J gene usage.
The number next to each leaf refers to the sequence count. A triangle
shaped leaf indicates the most frequent sequence. The distance between
leaves on the horizontal axis corresponds to the sequence similarity
(i.e. the further apart the leaves are horizontally, the less similar
the sequences are to one another).
nuc_IGH_table <- LymphoSeq2::productiveSeq(study_table = IGH_table, aggregate = "junction")
LymphoSeq2::phyloTree(study_table = nuc_IGH_table,
repertoire_ids = "IGH_MVQ92552A_BL",
type = "junction",
layout = "rectangular")
Warning:
[1m
[22mThe `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as
of ggplot2 3.3.4.
[36mℹ
[39m The deprecated feature was likely used in the
[34mLymphoSeq2
[39m package.
Please report the issue at
[3m
[34m<https://github.com/shashidhar22/LymphoSeq2/issues>
[39m
[23m.
[90mThis warning is displayed once every 8 hours.
[39m
[90mCall `lifecycle::last_lifecycle_warnings()` to see where this warning was
[39m
[90mgenerated.
[39m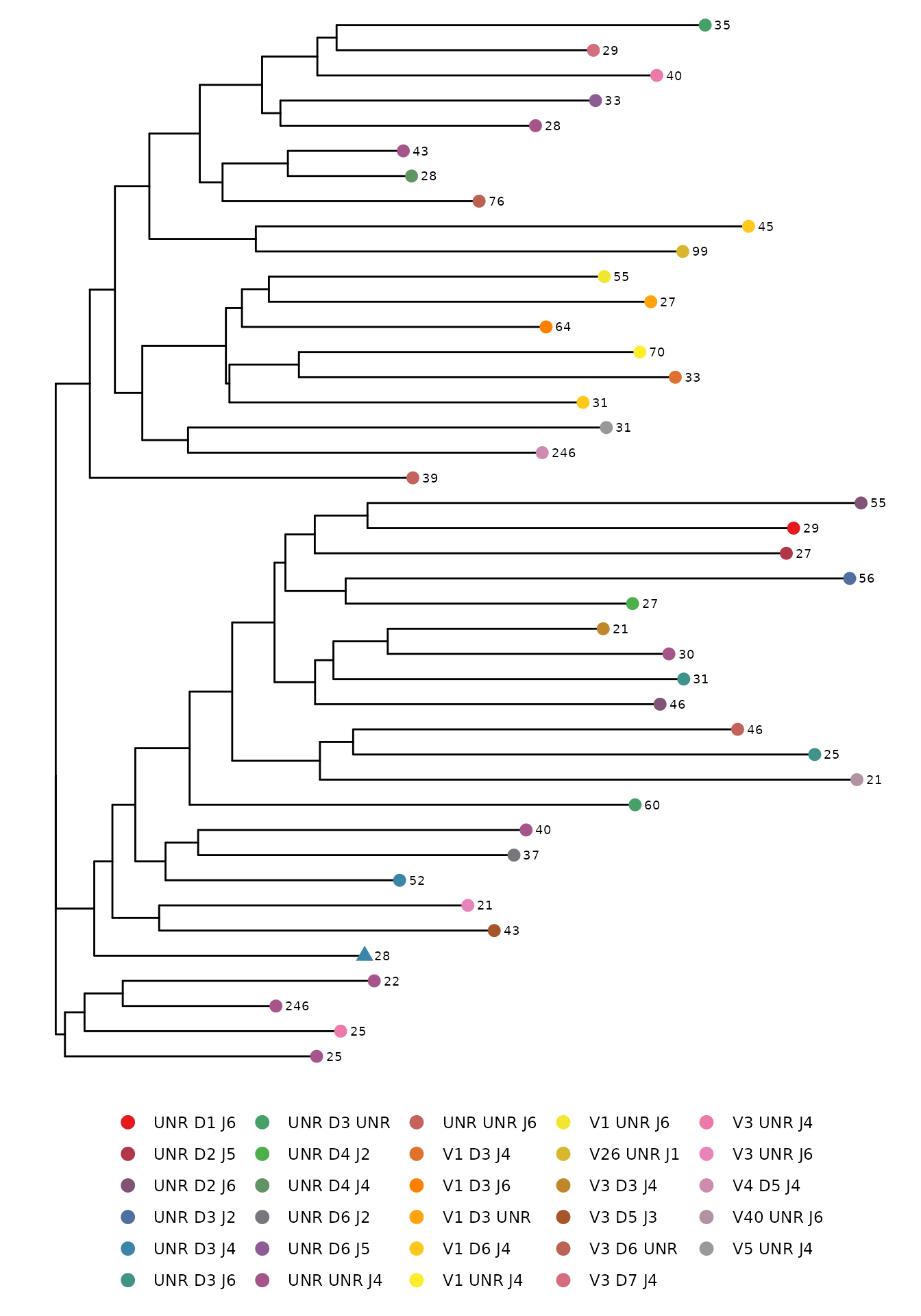
Multiple sequence alignment
In LymphoSeq2, you can perform a multiple sequence alignment using
one of three methods provided by the Bioconductor msa package (ClustalW,
ClustalOmega, or Muscle), the change in functionality however is, now
the function returns a msa S4 object. One may perform the
alignment of all amino acid or nucleotide sequences in a single sample.
Alternatively, one may search for a given sequence within a list of
samples using an edit distance threshold.
alignment <- LymphoSeq2::alignSeq(study_table = nuc_IGH_table,
repertoire_ids = "IGH_MVQ92552A_BL",
type = "junction_aa",
method = "ClustalW")use default substitution matrix
LymphoSeq2::plotAlignment(alignment)
#> Coordinate system already present. Adding new coordinate system, which will
#> replace the existing one.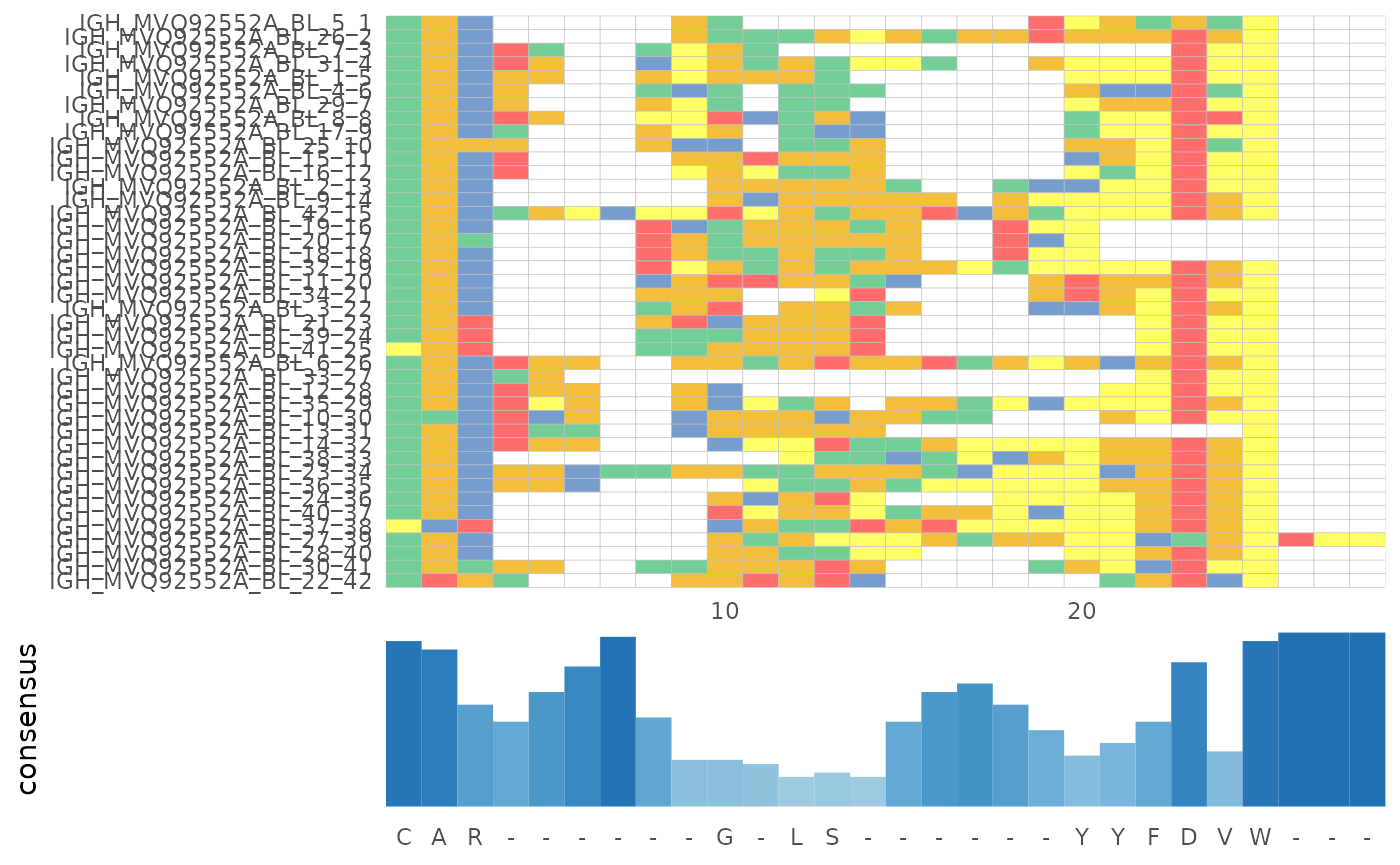
Searching for sequences
To search for one or more amino acid or nucleotide CDR3 sequences in
a list of data frames, use the function searchSeq. The
function allows sequence search with a edit distance threshold. For
example, an edit distance of 0 means the sequences are identical and an
edit distance of 1 indicates that the sequences differ by a single amino
acid or nucleotide. Match options include “global” matching which
performs end-to-end matching of sequences. “partial” matching allows
searching for sub strings with CDR3 sequences.
LymphoSeq2::searchSeq(study_table = aa_table,
sequence = "CASSPVSNEQFF",
seq_type = "junction_aa",
match = "global",
edit_distance = 0)
#> # A tibble: 1 × 13
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_Unsorted_0 CASSPVSNEQFF TRBV28-1 TRBD2-1 TRBJ2… TRBV28 TRBD2 TRBJ2
#> # ℹ 5 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>, edit_distance <dbl>, searchSequence <chr>Searching for published sequences
To search your entire list of data frames for a published amino acid
CDR3 TCRB sequence with known antigen specificity, use the function
searchPublished.
LymphoSeq2::searchPublished(study_table = aa_table) %>%
dplyr::filter(!is.na(PMID))
#> # A tibble: 26 × 16
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_CD4_949 CASSQDPGYE… TRBV4… TRBD1… TRBJ2… TRBV4 TRBD1 TRBJ2
#> 2 TRB_CD8_949 CASSPGTGTY… TRBV1… TRBD1… TRBJ1… TRBV10 TRBD1 TRBJ1
#> 3 TRB_CD8_949 CASSPSRNTE… TRBV4… TRBD2… TRBJ1… TRBV4 TRBD2 TRBJ1
#> 4 TRB_CD8_949 CASSYSGNTE… NA NA TRBJ1… NA NA TRBJ1
#> 5 TRB_CD8_CMV_369 CASSPARNTE… TRBV4… NA TRBJ1… TRBV4 NA TRBJ1
#> 6 TRB_CD8_CMV_369 CASSPGTGTY… TRBV1… TRBD1… TRBJ1… TRBV10 TRBD1 TRBJ1
#> 7 TRB_CD8_CMV_369 CASSPSRNTE… TRBV4… TRBD2… TRBJ1… TRBV4 TRBD2 TRBJ1
#> 8 TRB_CD8_CMV_369 CASSYSGNTE… NA NA TRBJ1… NA NA TRBJ1
#> 9 TRB_Unsorted_0 CASSPQRNTE… TRBV4… TRBD2… TRBJ1… TRBV4 TRBD2 TRBJ1
#> 10 TRB_Unsorted_1320 CASSLEGDQP… TRBV5… TRBD1… TRBJ1… TRBV5 TRBD1 TRBJ1
#> # ℹ 16 more rows
#> # ℹ 8 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>, PMID <fct>, HLA <fct>, antigen <fct>,
#> # epitope <fct>, prevalence <dbl>For each found sequence, a table is provides listing the antigen, epitope, HLA type, PubMed ID (PMID), and prevalence percentage of the sequence among 55 healthy donor blood samples.
You can even search of productive CDR3 amino acid sequences from the
repertoires that are found in public databases such as VdjDB, IEDB, and
McPas-TCR using the function searchDB. By specifying
dbname="all" searchDB will look for each CDR3
amino acid sequence in the dataset in all three public databases. You
can also pass a vector with any of the three databases (“VdjDB”, “IEDB”,
“McPAS-TCR”) to search just those databases.
LymphoSeq2::searchDB(study_table = aa_table, dbname = "all", chain = "trb")
#> # A tibble: 839 × 26
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_CD4_949 CAISVGGSSPLHF TRBV1… TRBD2… TRBJ1… TRBV10 TRBD2 TRBJ1
#> 2 TRB_CD4_949 CASDGGFRNTIYF TRBV1… TRBD2… TRBJ1… TRBV19 TRBD2 TRBJ1
#> 3 TRB_CD4_949 CASGGLNTEAFF NA NA TRBJ1… NA NA TRBJ1
#> 4 TRB_CD4_949 CASGLVAGSTLGGE… TRBV1… TRBD2… TRBJ2… TRBV12 TRBD2 TRBJ2
#> 5 TRB_CD4_949 CASGTGGETQYF TRBV6… TRBD2… TRBJ2… TRBV6 TRBD2 TRBJ2
#> 6 TRB_CD4_949 CASHSSGNTIYF TRBV6… NA TRBJ1… TRBV6 NA TRBJ1
#> 7 TRB_CD4_949 CASKPPGQGGYGYTF TRBV6… TRBD1… TRBJ1… TRBV6 TRBD1 TRBJ1
#> 8 TRB_CD4_949 CASMIDPSGNTIYF TRBV5… NA TRBJ1… TRBV5 NA TRBJ1
#> 9 TRB_CD4_949 CASNARVDSPLHF TRBV6… TRBD1… TRBJ1… TRBV6 TRBD1 TRBJ1
#> 10 TRB_CD4_949 CASRLGESPLHF NA NA TRBJ1… NA NA TRBJ1
#> # ℹ 829 more rows
#> # ℹ 18 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>, tra_cdr3_aa <chr>, gene <chr>, epitope <chr>,
#> # pathology <chr>, antigen <chr>, tra_v_call <chr>, tra_j_call <chr>,
#> # mhc_allele <chr>, reference <chr>, score <dbl>, cell_type <chr>,
#> # source <chr>, trb_v_call <chr>, trb_j_call <chr>, Species <chr>Visualizing repertoire diversity
Antigen receptor repertoire diversity can be characterized by a
number such as clonality or Gini coefficient calculated by the
clonality function. Alternatively, you can visualize the
repertoire diversity by plotting the Lorenz curve for each sample as
defined above. In this plot, the more diverse samples will appear near
the dotted diagonal line (the line of equality) whereas the more clonal
samples will appear to have a more bowed shape.
samples <- aa_table %>%
dplyr::pull(repertoire_id) %>% unique()
LymphoSeq2::lorenzCurve(repertoire_ids = samples, study_table = aa_table)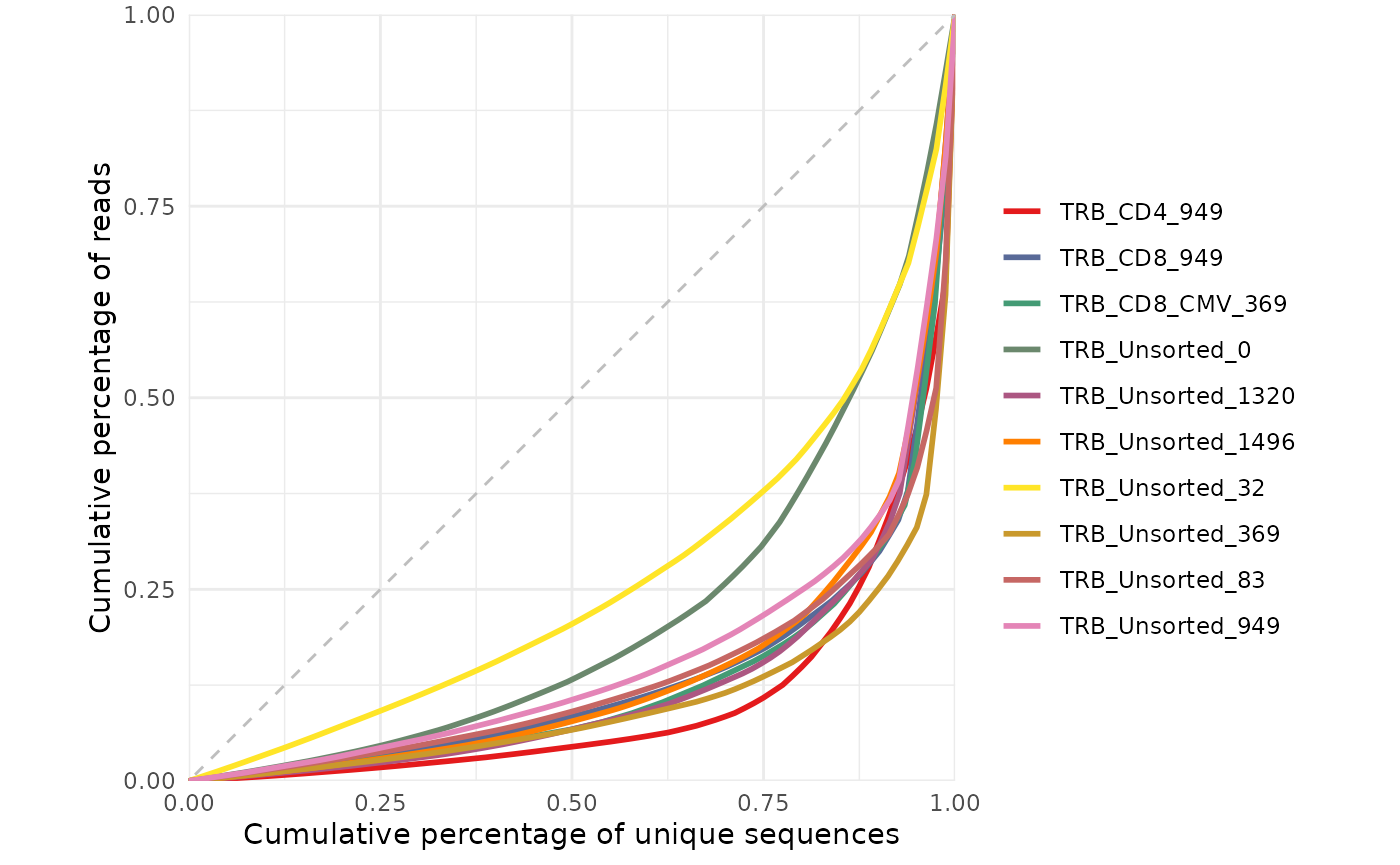
Alternatively, you can get a feel for the repertoire diversity by
plotting the cumulative frequency of a selected number of the top most
frequent clones using the function topSeqsPlot. In this
case, each of the top sequences are represented by a different color and
all less frequent clones will be assigned a single color (violet).
LymphoSeq2::topSeqsPlot(study_table = aa_table, top = 10)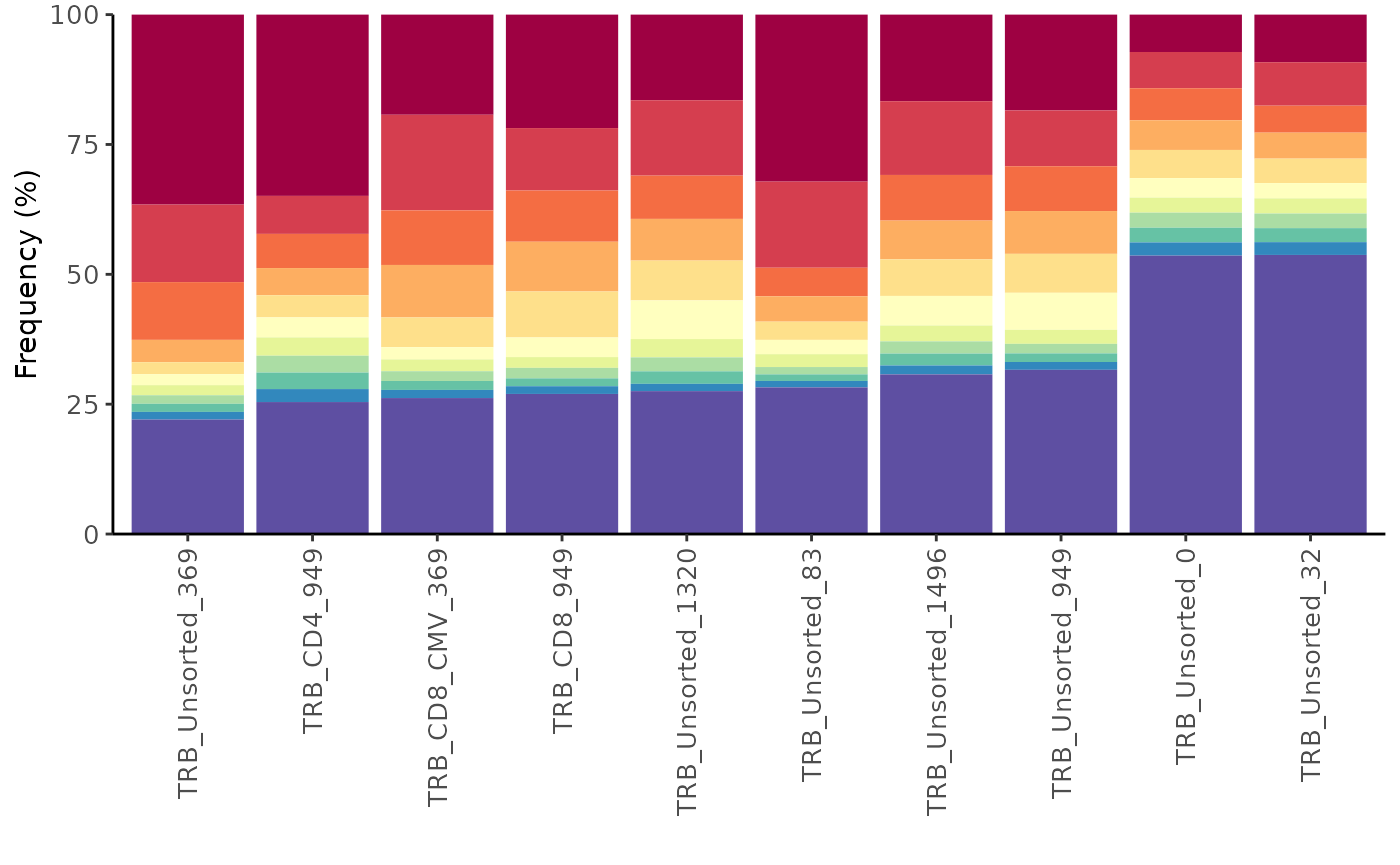
Both of these functions are built using the ggplot2 package. You can
reformat the plot using ggplot2 functions. Please refer to the
lorenzCurve and topSeqsPlot manual for
specific examples.
Comparing samples
To compare the T or B cell repertoires of all samples in a pairwise
fashion, use the bhattacharyyaMatrix or
similarityMatrix functions. Both the Bhattacharyya
coefficient and similarity score are measures of the amount of overlap
between two samples. The value for each ranges from 0 to 1 where 1
indicates the sequence frequencies are identical in the two samples and
0 indicates no shared frequencies. The Bhattacharyya coefficient differs
from the similarity score in that it involves weighting each shared
sequence in the two distributions by the arithmetic mean of the
frequency of each sequence, while calculating the similarity scores
involves weighting each shared sequence in the two distributions by the
geometric mean of the frequency of each sequence in the two
distributions.
bhattacharyya_matrix <- LymphoSeq2::scoringMatrix(aa_table, mode = "Bhattacharyya")
LymphoSeq2::pairwisePlot(bhattacharyya_matrix)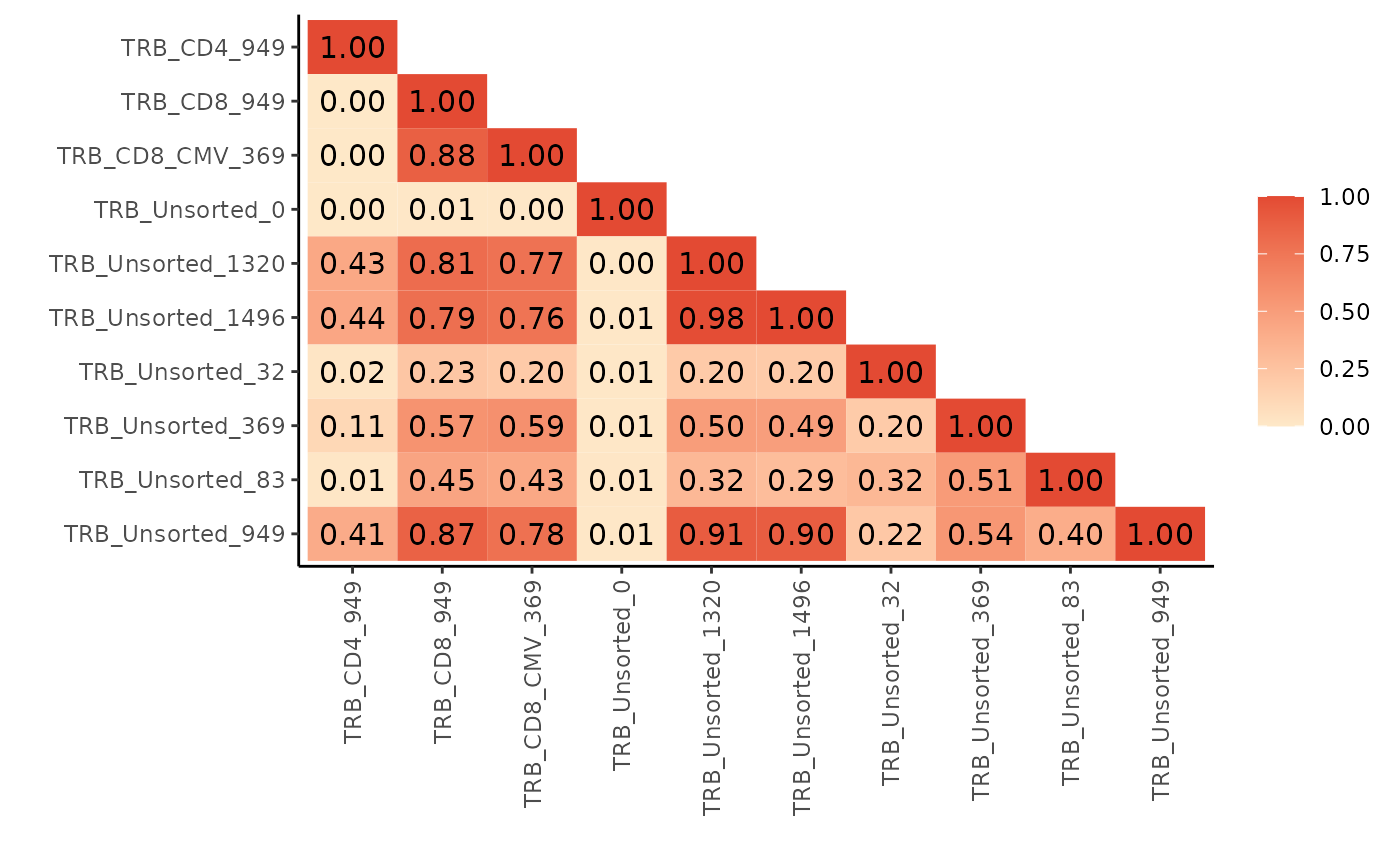
To view sequences shared between two or more samples, use the
function commonSeqs. This function requires that a
productive amino acid list be specified.
common <- LymphoSeq2::commonSeqs(study_table = aa_table,
repertoire_ids = c("TRB_Unsorted_0", "TRB_Unsorted_32"))
common
#> # A tibble: 1 × 3
#> junction_aa TRB_Unsorted_0 TRB_Unsorted_32
#> <chr> <dbl> <dbl>
#> 1 CASSQDRTGQYGYTF 0.00429 0.0152To visualize the number of overlapping sequences between two or three
samples in the form of a Venn diagram, use the function
commonSeqVenn
LymphoSeq2::commonSeqsVenn(repertoire_ids = c("TRB_Unsorted_32", "TRB_Unsorted_83"),
amino_table = aa_table)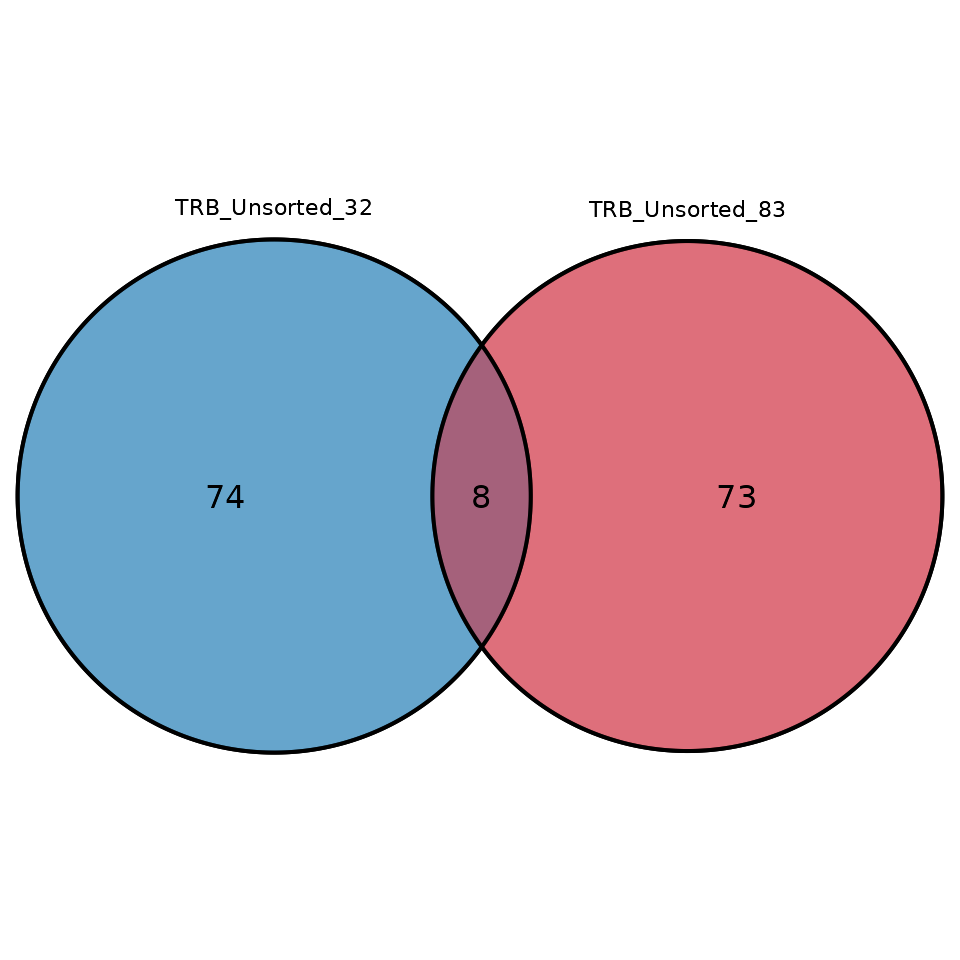
LymphoSeq2::commonSeqsVenn(repertoire_ids = c("TRB_Unsorted_0", "TRB_Unsorted_32", "TRB_Unsorted_83"),
amino_table = aa_table)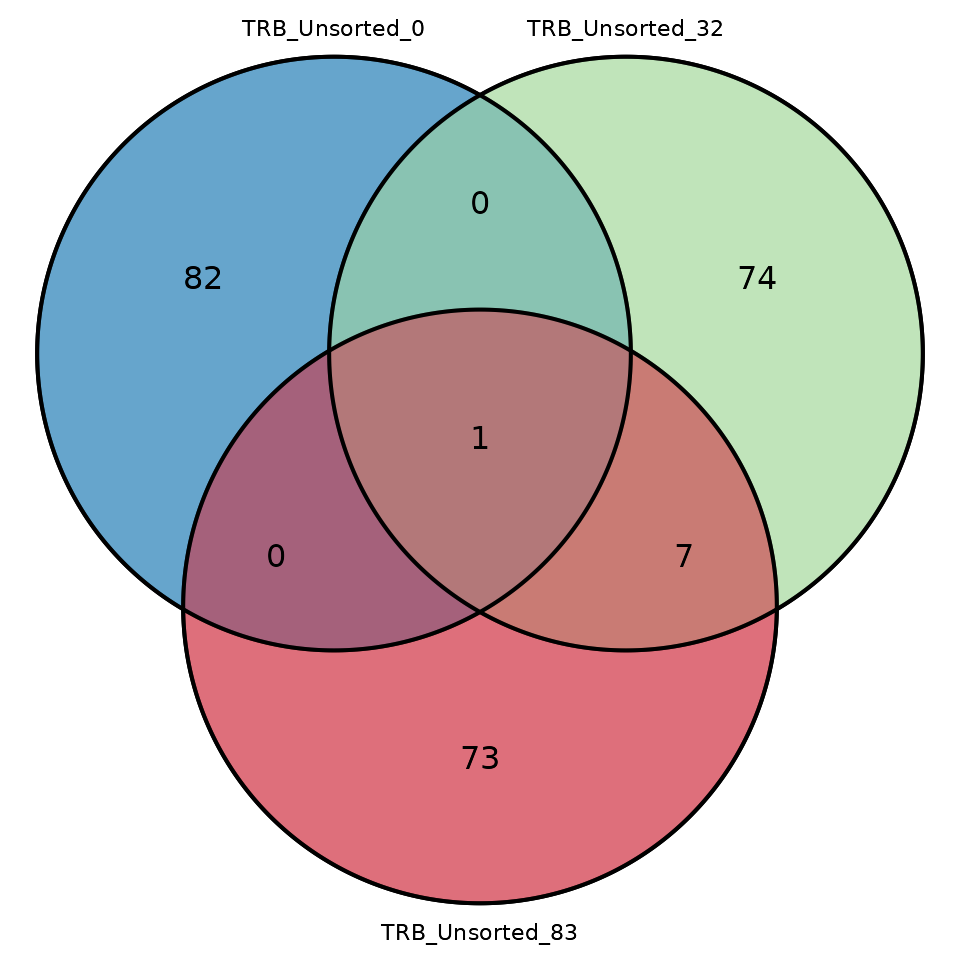
To compare the frequency of sequences between two samples as a
scatter plot, use the function commonSeqsPlot.
LymphoSeq2::commonSeqsPlot("TRB_Unsorted_32", "TRB_Unsorted_83",
amino_table = aa_table, show = "common")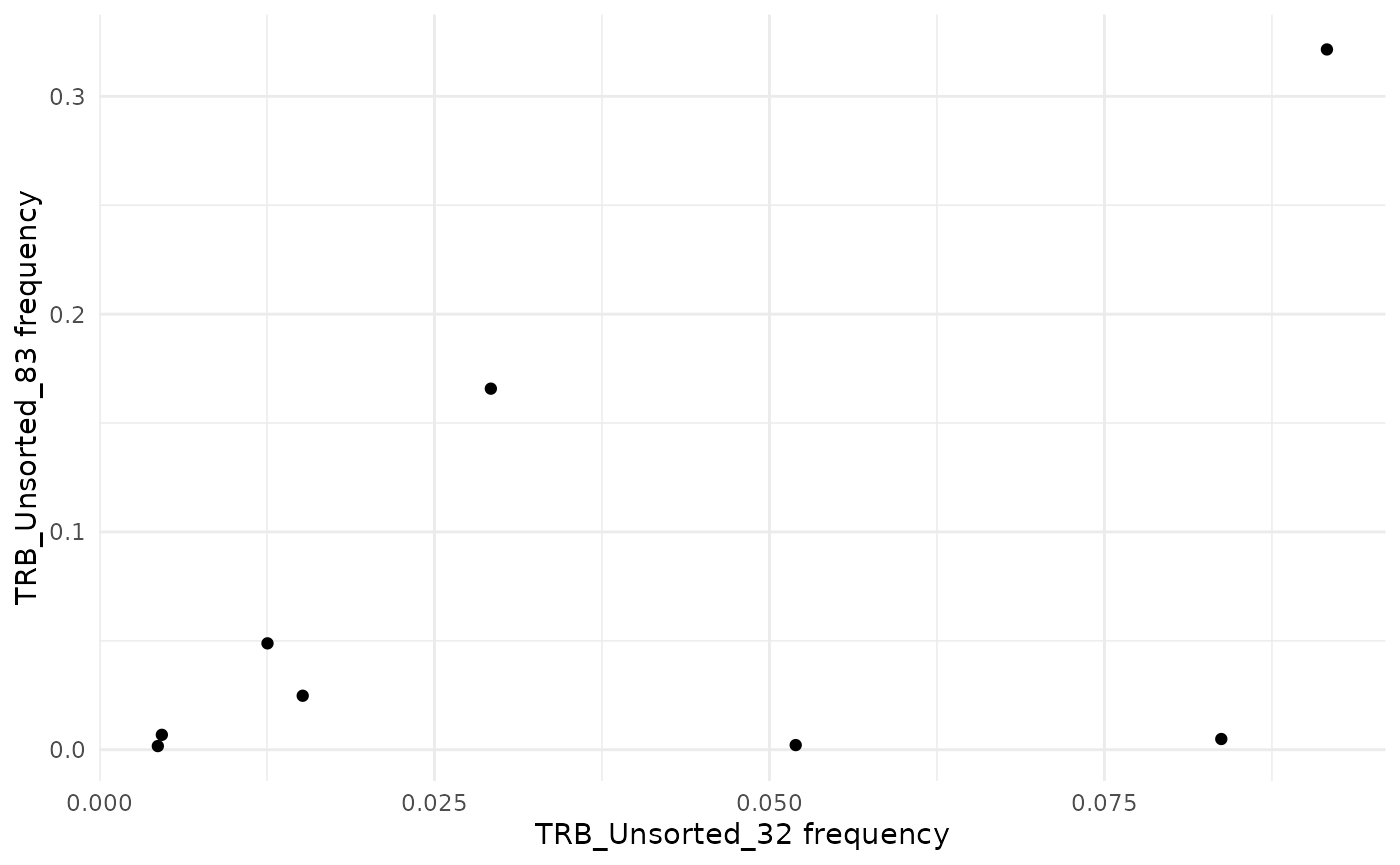
If you have more than 3 samples to compare, use the
commonSeqBar function. You can chose to color a single
sample with the color.sample argument or a desired
intersection with the color.intersection argument.
LymphoSeq2::commonSeqsBar(amino_table = aa_table,
repertoire_ids = c("TRB_CD4_949", "TRB_CD8_949",
"TRB_Unsorted_949", "TRB_Unsorted_1320"),
color_sample = "TRB_CD8_949",
labels = "no")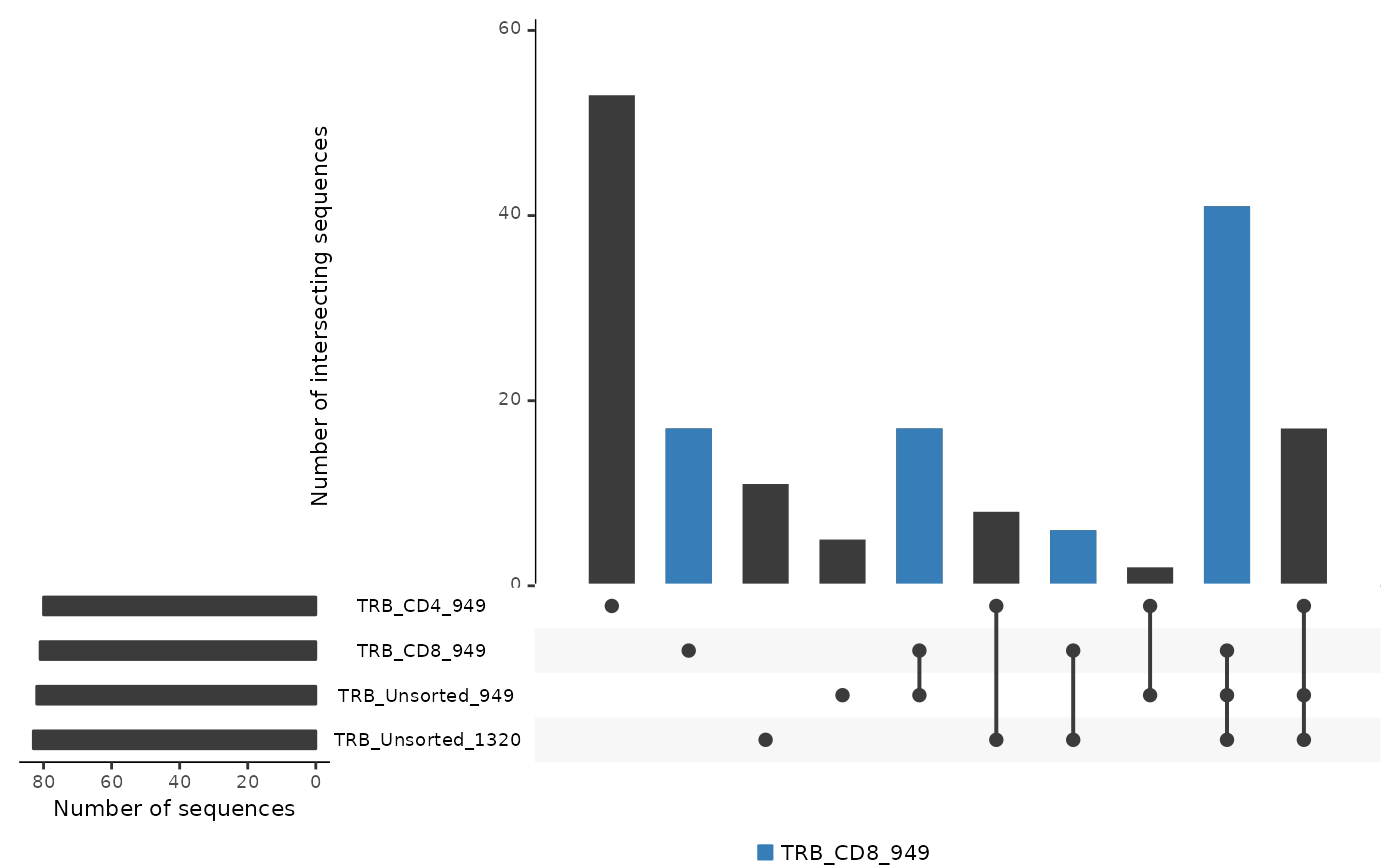
Differential abundance
When comparing a sample from two different time points, it is useful
to identify sequences that are significantly more or less abundant in
one versus the other time point (DeWitt, W.S., et al. Journal of
Virology 2015 89(8):4517-4526). The differentialAbundance
function uses a Fisher exact test to calculate differential abundance of
each sequence in two time points and reports the log2 transformed fold
change, P value and adjusted P value.
LymphoSeq2::differentialAbundance(study_table = aa_table,
repertoire_ids =c("TRB_Unsorted_949",
"TRB_Unsorted_1320"),
type = "junction_aa", q = 0.01)
#> # A tibble: 107 × 6
#> junction_aa TRB_Unsorted_949 TRB_Unsorted_1320 p q l2fc
#> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
#> 1 CAIKMETPNGEQYF 29 326 1.14e- 6 1.14e- 6 -3.49
#> 2 CAISEGQGVKPQHF 0 167 1.09e- 2 1.09e- 2 -Inf
#> 3 CAISESGVLNEKLFF 13 150 1.20e- 3 1.20e- 3 -3.53
#> 4 CASDGGFRNTIYF 17 387 1.40e- 1 1.40e- 1 -4.51
#> 5 CASKPPGQGGYGYTF 0 173 1.12e- 2 1.12e- 2 -Inf
#> 6 CASNRVPEETQYF 0 127 5.75e- 2 5.75e- 2 -Inf
#> 7 CASNSKADSTDTQYF 21 1325 1.52e- 3 1.52e- 3 -5.98
#> 8 CASRDGQGSGNTIYF 48 358 6.73e-16 6.73e-16 -2.90
#> 9 CASREDRGSSPLHF 0 147 2.45e- 2 2.45e- 2 -Inf
#> 10 CASRLGPGAGDEAFF 12 619 1.26e- 1 1.26e- 1 -5.69
#> # ℹ 97 more rowsFinding recurring sequences
To create a tibble of unique, productive amino acid sequences as rows
and sample names as headers use the seqMatrix function.
Each value in the data frame represents the frequency that each sequence
appears in the sample. You can specify your own list of sequences or all
unique sequences in the list using the output of the function
uniqueSeqs. The uniqueSeqs function creates a
tibble of all unique, productive sequences and reports the total count
in all samples.
unique_seqs <- LymphoSeq2::uniqueSeqs(productive_table = aa_table)
unique_seqs
#> # A tibble: 438 × 2
#> junction_aa duplicate_count
#> <chr> <dbl>
#> 1 CASSQDWERLGEQFF 99480
#> 2 CASSLQGREKLFF 90563
#> 3 CASSQDLMTVDSLFAGANVLTF 68679
#> 4 CASSPAGAYYNEQFF 30418
#> 5 CASSPPTGERDTQYF 24552
#> 6 CASSLAGDSQETQYF 22147
#> 7 CASSESAGSTGELFF 17438
#> 8 CASRDGQGSGNTIYF 11516
#> 9 CASSPSRNTEAFF 8705
#> 10 CASSQDRTGQYGYTF 8017
#> # ℹ 428 more rows
sequence_matrix <- LymphoSeq2::seqMatrix(amino_table = aa_table, sequences = unique_seqs$junction_aa)
sequence_matrix
#> # A tibble: 438 × 11
#> junction_aa TRB_CD4_949 TRB_CD8_949 TRB_CD8_CMV_369 TRB_Unsorted_0
#> <chr> <dbl> <dbl> <dbl> <dbl>
#> 1 CAISVGGSSPLHF 0.000695 0 0 0
#> 2 CASDGGFRNTIYF 0.0318 0 0 0
#> 3 CASGGLNTEAFF 0.00160 0 0 0
#> 4 CASGLVAGSTLGGETQYF 0.00202 0 0 0
#> 5 CASGTGGETQYF 0.00146 0 0 0
#> 6 CASHSSGNTIYF 0.000765 0 0 0
#> 7 CASKPPGQGGYGYTF 0.00827 0 0 0
#> 8 CASMIDPSGNTIYF 0.000765 0 0 0
#> 9 CASNARVDSPLHF 0.000834 0 0 0
#> 10 CASRLGESPLHF 0.00167 0 0 0
#> # ℹ 428 more rows
#> # ℹ 6 more variables: TRB_Unsorted_1320 <dbl>, TRB_Unsorted_1496 <dbl>,
#> # TRB_Unsorted_32 <dbl>, TRB_Unsorted_369 <dbl>, TRB_Unsorted_83 <dbl>,
#> # TRB_Unsorted_949 <dbl>If just the top clones with a frequency greater than a specified
amount are of interest to you, then use the topFreq
function. This creates a tibble of the top productive amino acid
sequences having a minimum specified frequency and reports the minimum,
maximum, and mean frequency that the sequence appears in a list of
samples. For TCRB sequences, the prevalence percentage and the published
antigen specificity of that sequence are also provided.
top_freq <- LymphoSeq2::topFreq(productive_table = aa_table, frequency = 0.001)
top_freq
#> # A tibble: 425 × 7
#> junction_aa minFrequency maxFrequency meanFrequency numberSamples prevalence
#> <chr> <dbl> <dbl> <dbl> <int> <dbl>
#> 1 CASSQDRTGQY… 0.00429 0.0248 0.0106 9 0
#> 2 CASSLQGREKL… 0.0569 0.322 0.113 8 0
#> 3 CASSQDLMTVD… 0.0292 0.166 0.0924 8 0
#> 4 CASSREGDQPQ… 0.00157 0.0520 0.00913 8 0
#> 5 CASRDGQGSGN… 0.00278 0.0351 0.0155 7 0
#> 6 CASSPFDRGPD… 0.00508 0.0165 0.0112 7 0
#> 7 CASSQDLGQAF… 0.00223 0.0235 0.0111 7 0
#> 8 CASSQDSSDTE… 0.00147 0.0488 0.0106 7 0
#> 9 CAIKMETPNGE… 0.00253 0.0118 0.00704 7 0
#> 10 CASSPGTGTYG… 0.00121 0.0156 0.00585 7 0
#> # ℹ 415 more rows
#> # ℹ 1 more variable: antigen <fct>One very useful thing to do is merge the output of
seqMatrix and topFreq.
top_freq_matrix <- dplyr::full_join(top_freq, sequence_matrix)
#> Joining with `by = join_by(junction_aa)`
top_freq_matrix
#> # A tibble: 438 × 17
#> junction_aa minFrequency maxFrequency meanFrequency numberSamples prevalence
#> <chr> <dbl> <dbl> <dbl> <int> <dbl>
#> 1 CASSQDRTGQY… 0.00429 0.0248 0.0106 9 0
#> 2 CASSLQGREKL… 0.0569 0.322 0.113 8 0
#> 3 CASSQDLMTVD… 0.0292 0.166 0.0924 8 0
#> 4 CASSREGDQPQ… 0.00157 0.0520 0.00913 8 0
#> 5 CASRDGQGSGN… 0.00278 0.0351 0.0155 7 0
#> 6 CASSPFDRGPD… 0.00508 0.0165 0.0112 7 0
#> 7 CASSQDLGQAF… 0.00223 0.0235 0.0111 7 0
#> 8 CASSQDSSDTE… 0.00147 0.0488 0.0106 7 0
#> 9 CAIKMETPNGE… 0.00253 0.0118 0.00704 7 0
#> 10 CASSPGTGTYG… 0.00121 0.0156 0.00585 7 0
#> # ℹ 428 more rows
#> # ℹ 11 more variables: antigen <fct>, TRB_CD4_949 <dbl>, TRB_CD8_949 <dbl>,
#> # TRB_CD8_CMV_369 <dbl>, TRB_Unsorted_0 <dbl>, TRB_Unsorted_1320 <dbl>,
#> # TRB_Unsorted_1496 <dbl>, TRB_Unsorted_32 <dbl>, TRB_Unsorted_369 <dbl>,
#> # TRB_Unsorted_83 <dbl>, TRB_Unsorted_949 <dbl>Tracking sequences across samples
To visually track the frequency of sequences across multiple samples,
use the function cloneTrack This function takes the output
from the seqMatrix function. You can specify a character
vector of amino acid sequences using the parameter track to highlight
those sequences with a different color. Alternatively, you can highlight
all of the sequences from a given sample using the parameter map. If the
mapping feature is use, then you must specify a productive amino acid
list and a character vector of labels to title the mapped samples.
ctable <- LymphoSeq2::cloneTrack(study_table = aa_table,
sample_list = c("TRB_CD8_949", "TRB_CD8_CMV_369"))
LymphoSeq2::plotTrack(ctable)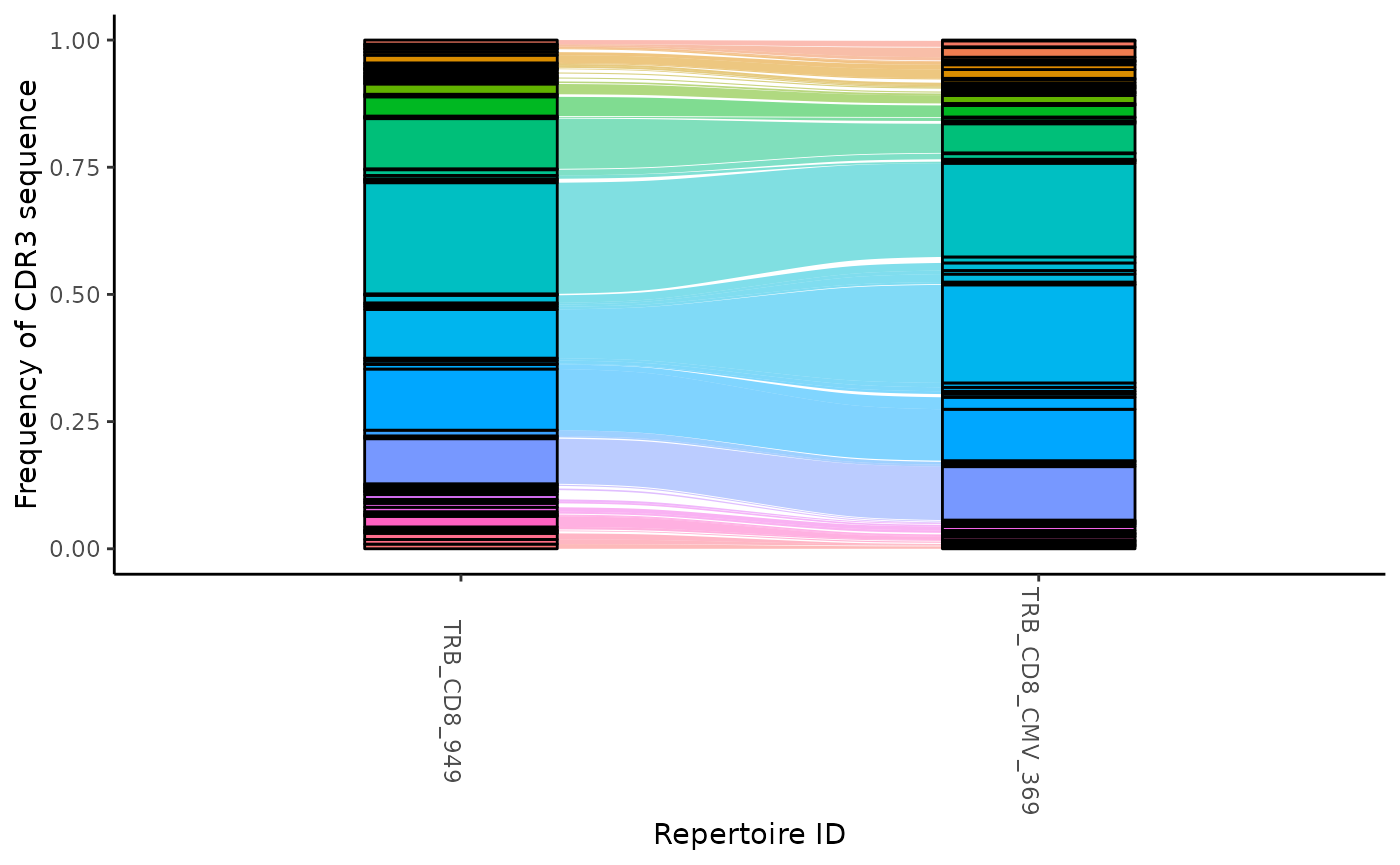
You can track particular sequences across samples by providing an optional list of CDR3 amino acid sequences.
ttable <- LymphoSeq2::topSeqs(aa_table, top = 10)
ctable <- LymphoSeq2::cloneTrack(ttable)
LymphoSeq2::plotTrack(ctable, alist = c("CASSESAGSTGELFF", "CASSLAGDSQETQYF")) + ggplot2::theme(legend.position = "bottom")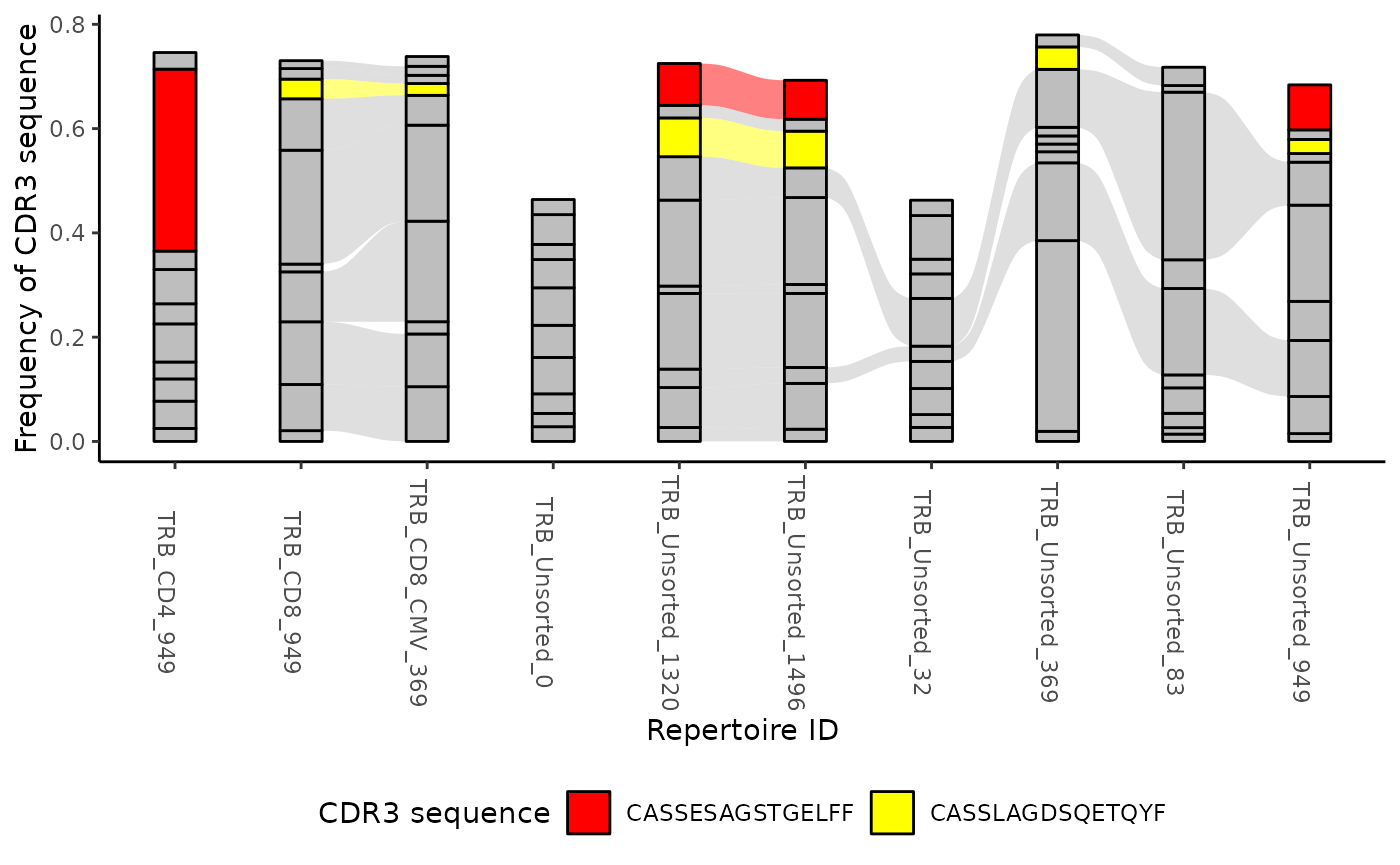
Alternatively you can use the function plotTrackSingular
to retrieve a list of alluvial diagrams each tracking one single amino
acid from the clone track table. Considering that a plot is generated
for each unique CDR3 sequence, we recommend running this feature on a
clone track table derived from only the top sequences from each
repertoire as described in the example above.
lalluvial <- ctable %>% LymphoSeq2::topSeqs(top = 1) %>%
LymphoSeq2::plotTrackSingular()
lalluvial[[1]]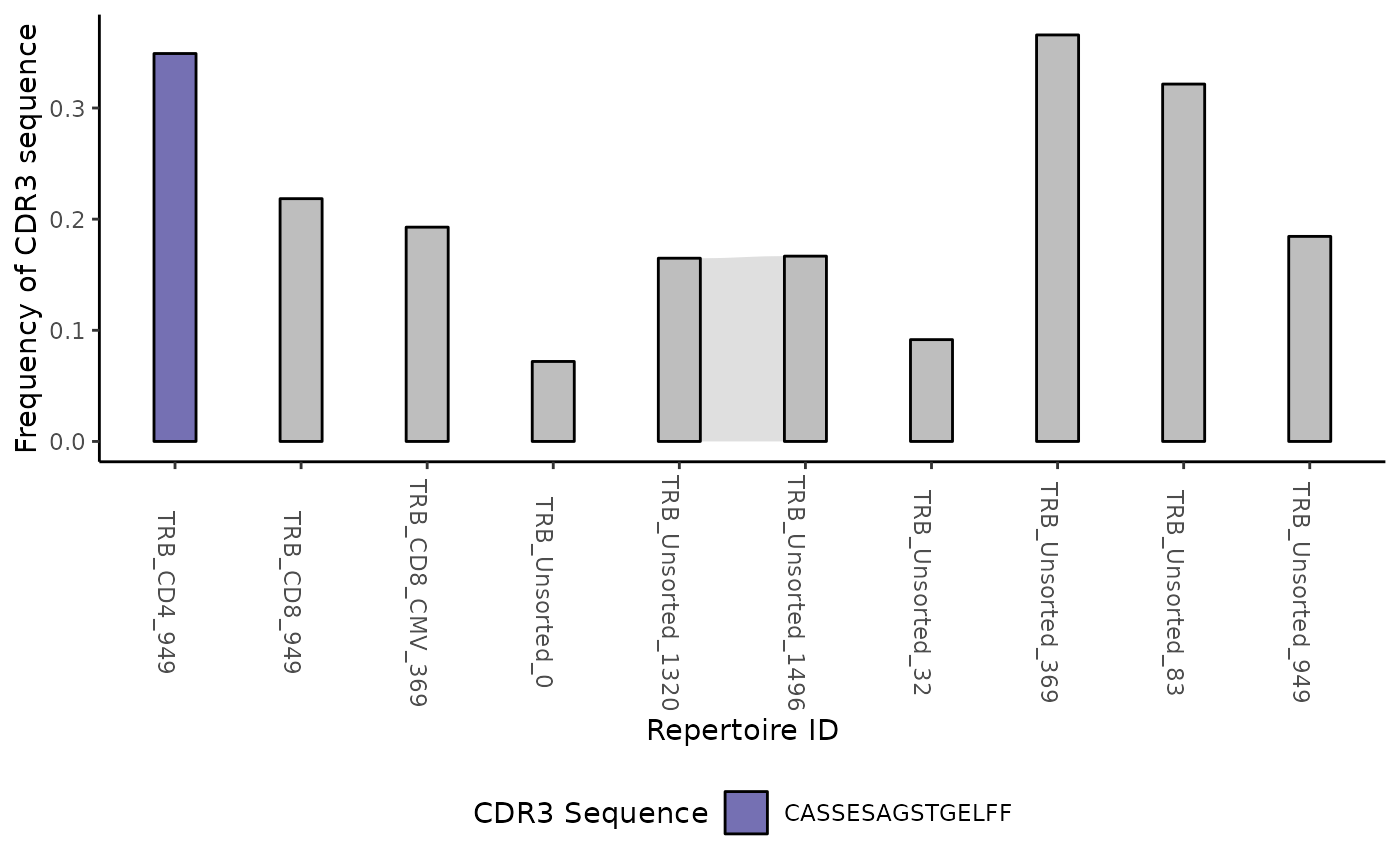
Comparing V(D)J gene usage
To compare the V, D, and J gene usage across samples, start by
creating a data frame of V, D, and J gene counts and frequencies using
the function geneFreq. You can specify if you are
interested in the “VDJ”, “DJ”, “VJ”, “DJ”, “V”, “D”, or “J” loci using
the locus parameter. Set family to TRUE if you prefer the family names
instead of the gene names as reported by ImmunoSeq.
vGenes <- LymphoSeq2::geneFreq(nuc_table, locus = "V", family = TRUE)
vGenes
#> # A tibble: 167 × 5
#> repertoire_id gene_name duplicate_count gene_type gene_frequency
#> <chr> <chr> <dbl> <chr> <dbl>
#> 1 TRB_CD4_949 NA 2945 v_family 0.205
#> 2 TRB_CD4_949 TRBV10 5071 v_family 0.353
#> 3 TRB_CD4_949 TRBV11 107 v_family 0.00744
#> 4 TRB_CD4_949 TRBV12 29 v_family 0.00202
#> 5 TRB_CD4_949 TRBV18 226 v_family 0.0157
#> 6 TRB_CD4_949 TRBV19 1643 v_family 0.114
#> 7 TRB_CD4_949 TRBV2 230 v_family 0.0160
#> 8 TRB_CD4_949 TRBV21 18 v_family 0.00125
#> 9 TRB_CD4_949 TRBV27 84 v_family 0.00584
#> 10 TRB_CD4_949 TRBV28 208 v_family 0.0145
#> # ℹ 157 more rowsTo create a chord diagram showing VJ or DJ gene associations from one
or more more samples, combine the output of geneFreq with
the function chordDiagramVDJ. This function works well the
topSeqs function that creates a data frame of a selected
number of top productive sequences. In the example below, a chord
diagram is made showing the association between V and J genes of just
the single dominant clones in each sample. The size of the ribbons
connecting VJ genes correspond to the number of samples that have that
recombination event. The thicker the ribbon, the higher the frequency of
the recombination.
top_seqs <- LymphoSeq2::topSeqs(nuc_table, top = 1)
LymphoSeq2::chordDiagramVDJ(study_table = top_seqs,
association = "VJ",
colors = c("darkred", "navyblue"))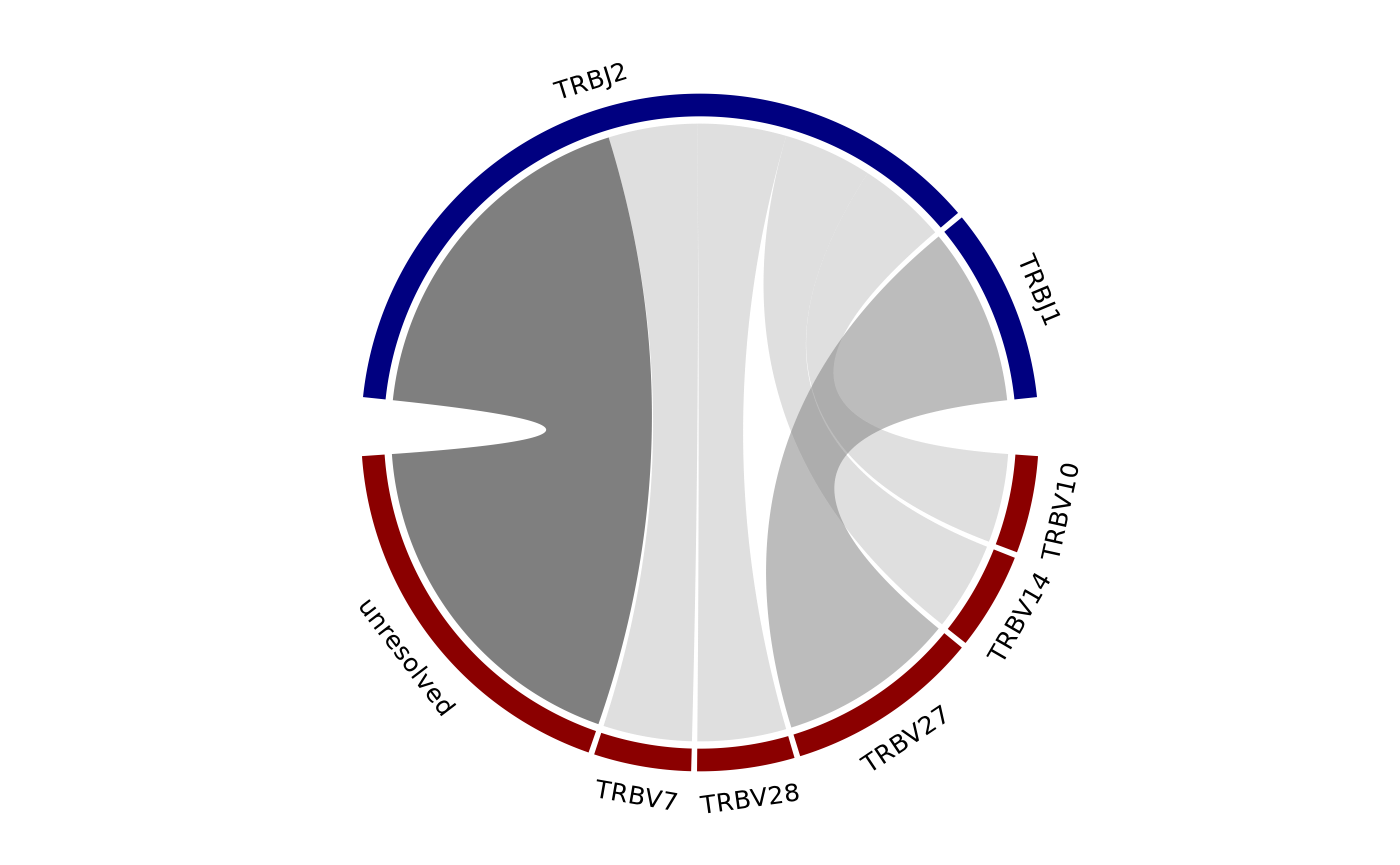
You can also visualize the results of geneFreq as a heat
map, word cloud, our cumulative frequency bar plot with the support of
additional R packages as shown below.
vGenes <- LymphoSeq2::geneFreq(nuc_table, locus = "V", family = TRUE)
RedBlue <- grDevices::colorRampPalette(rev(RColorBrewer::brewer.pal(11, "RdBu")))(256)
vtable <- vGenes %>% dplyr::filter(repertoire_id == "TRB_Unsorted_83") %>%
dplyr::select(gene_name, gene_frequency)
wordcloud2::wordcloud2(data = vtable,
color = RedBlue)
vGenes <- LymphoSeq2::geneFreq(nuc_table, locus = "V", family = TRUE) %>%
tidyr::pivot_wider(id_cols = gene_name,
names_from = repertoire_id,
values_from = gene_frequency,
values_fn = sum,
values_fill = 0)
gene_names <- vGenes %>%
dplyr::pull(gene_name)
vGenes <- vGenes %>%
dplyr::select(-gene_name) %>%
as.matrix()
rownames(vGenes) <- gene_names
pheatmap::pheatmap(vGenes, scale = "row")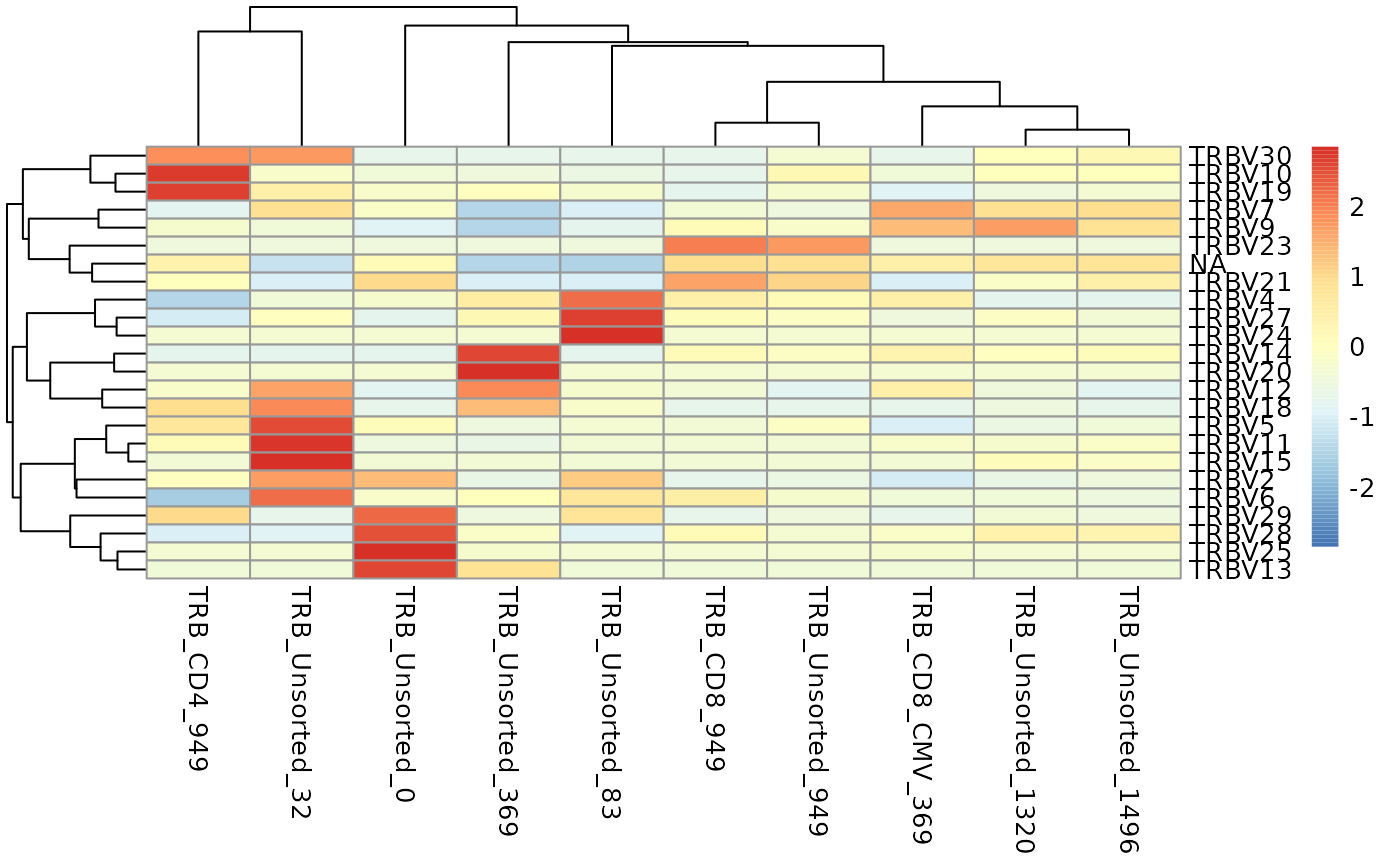
vGenes <- LymphoSeq2::geneFreq(nuc_table, locus = "V", family = TRUE)
multicolors <- grDevices::colorRampPalette(rev(RColorBrewer::brewer.pal(9, "Set1")))(28)
ggplot2::ggplot(vGenes, aes(x = repertoire_id, y = gene_frequency, fill = gene_name)) +
ggplot2::geom_bar(stat = "identity") +
ggplot2::theme_minimal() +
ggplot2::scale_y_continuous(expand = c(0, 0)) +
ggplot2::guides(fill = ggplot2::guide_legend(ncol = 2)) +
ggplot2::scale_fill_manual(values = multicolors) +
ggplot2::labs(y = "Frequency (%)", x = "", fill = "") +
ggplot2::theme(axis.text.x = ggplot2::element_text(angle = 90, vjust = 0.5, hjust = 1))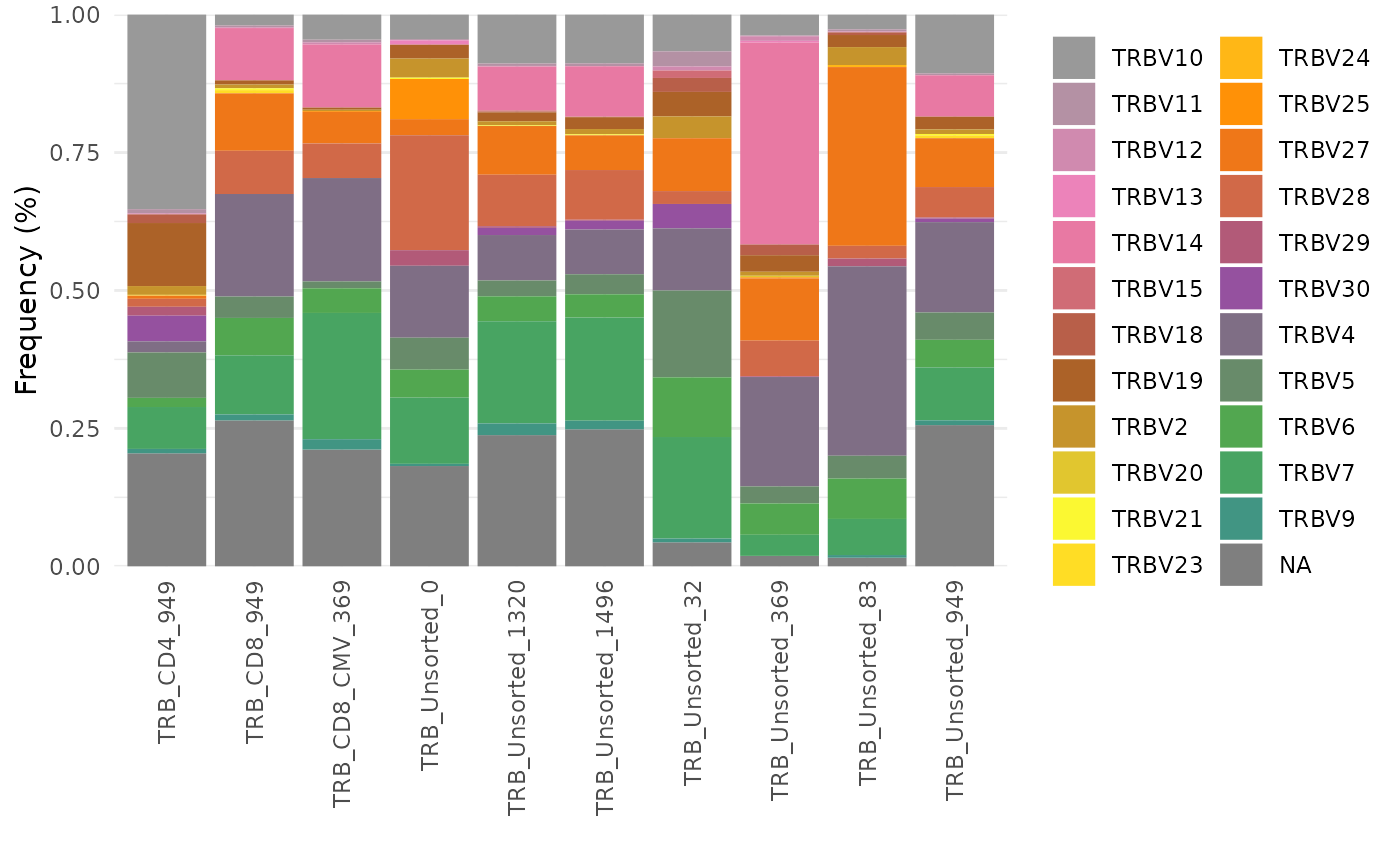
Removing sequences
Occasionally you may identify one or more sequences in your data set
that appear to be contamination. You can remove an amino acid sequence
from all data frames using the function removeSeq and
recompute frequencyCount for all remaining sequences.
LymphoSeq2::searchSeq(study_table = aa_table, sequence = "CASSESAGSTGELFF", seq_type = "junction_aa")
#> # A tibble: 4 × 13
#> repertoire_id junction_aa v_call d_call j_call v_family d_family j_family
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <chr>
#> 1 TRB_CD4_949 CASSESAGSTG… TRBV1… TRBD2… TRBJ2… TRBV10 TRBD2 TRBJ2
#> 2 TRB_Unsorted_1320 CASSESAGSTG… TRBV1… TRBD2… TRBJ2… TRBV10 TRBD2 TRBJ2
#> 3 TRB_Unsorted_1496 CASSESAGSTG… TRBV1… TRBD2… TRBJ2… TRBV10 TRBD2 TRBJ2
#> 4 TRB_Unsorted_949 CASSESAGSTG… TRBV1… TRBD2… TRBJ2… TRBV10 TRBD2 TRBJ2
#> # ℹ 5 more variables: reading_frame <chr>, duplicate_count <dbl>,
#> # duplicate_frequency <dbl>, edit_distance <dbl>, searchSequence <chr>
cleansed <- LymphoSeq2::removeSeq(study_table = aa_table, sequence = "CASSESAGSTGELFF")
LymphoSeq2::searchSeq(study_table = cleansed, sequence = "CASSESAGSTGELFF", seq_type = "junction_aa")
#> # A tibble: 0 × 13
#> # ℹ 13 variables: repertoire_id <chr>, junction_aa <chr>, v_call <chr>,
#> # d_call <chr>, j_call <chr>, v_family <chr>, d_family <chr>, j_family <chr>,
#> # reading_frame <chr>, duplicate_count <dbl>, duplicate_frequency <dbl>,
#> # edit_distance <dbl>, searchSequence <chr>Rarefaction curves
Rarefaction and extrapolation curves allow for comparison of TCR diversity across repertoires given a ideal sequencing depth. Rarefaction and extrapolation curves are drawn by sampling a sequencing dataset to various depths to understand the trajectory of sequence diversity and then extrapolating the curve to an ideal depth.
LymphoSeq2::plotRarefactionCurve(study_table = aa_table)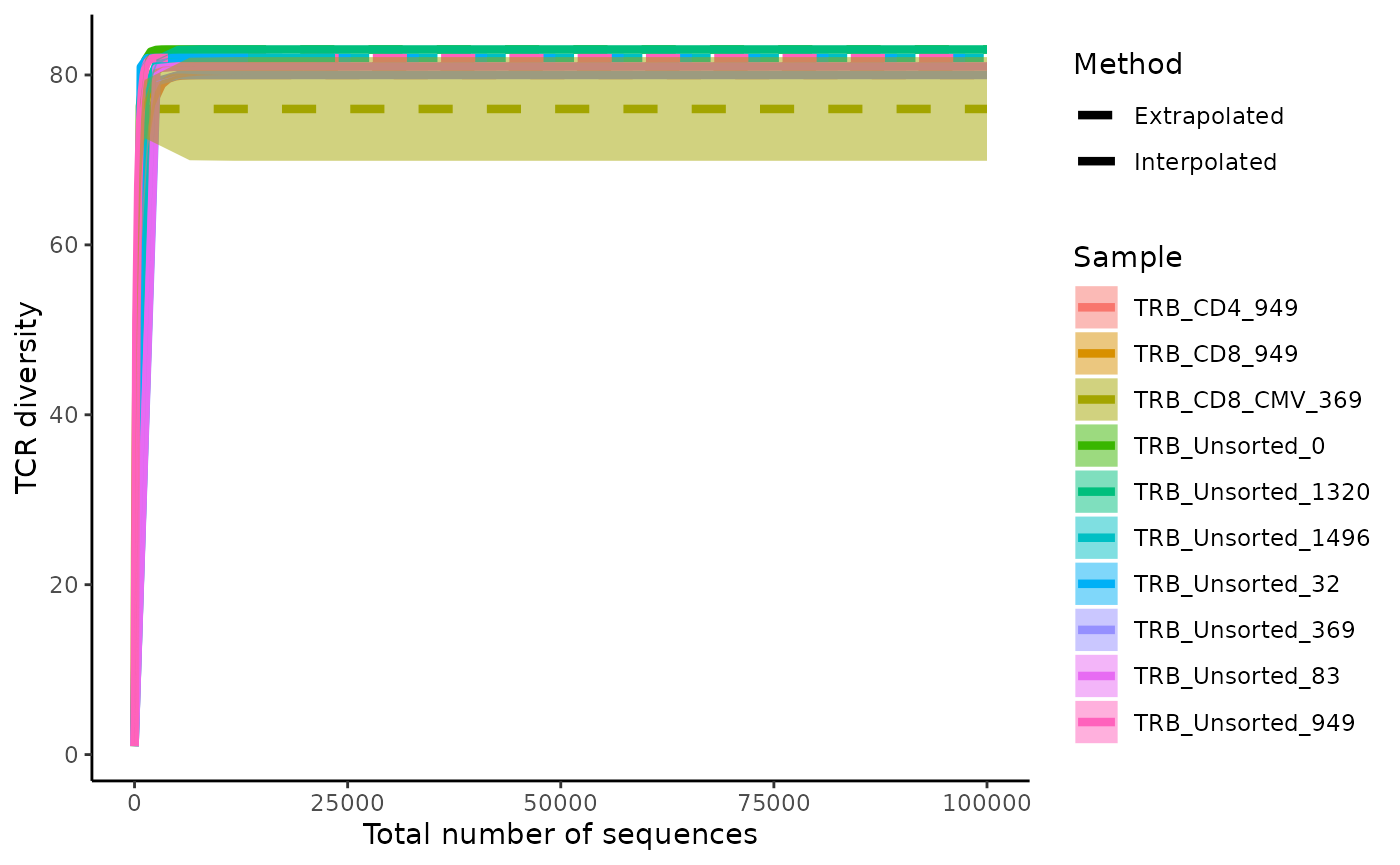
Session info
sessionInfo()
#> R version 4.3.0 (2023-04-21)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 22.04.2 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] vroom_1.6.3 lubridate_1.9.2 forcats_1.0.0 stringr_1.5.0
#> [5] dplyr_1.1.2 purrr_1.0.1 readr_2.1.4 tidyr_1.3.0
#> [9] tibble_3.2.1 ggplot2_3.4.2 tidyverse_2.0.0 wordcloud2_0.2.1
#> [13] RColorBrewer_1.1-3 LymphoSeq2_1.0.0 data.table_1.14.8
#>
#> loaded via a namespace (and not attached):
#> [1] bitops_1.0-7 gridExtra_2.3 formatR_1.14
#> [4] phangorn_2.11.1 rlang_1.1.1 magrittr_2.0.3
#> [7] compiler_4.3.0 reshape2_1.4.4 ggalluvial_0.12.5
#> [10] systemfonts_1.0.4 vctrs_0.6.2 maps_3.4.1
#> [13] quadprog_1.5-8 shape_1.4.6 pkgconfig_2.0.3
#> [16] crayon_1.5.2 fastmap_1.1.1 ellipsis_0.3.2
#> [19] XVector_0.40.0 labeling_0.4.2 utf8_1.2.3
#> [22] rmarkdown_2.22 tzdb_0.4.0 seqmagick_0.1.5
#> [25] UpSetR_1.4.0 ragg_1.2.5 bit_4.0.5
#> [28] xfun_0.39 zlibbioc_1.46.0 cachem_1.0.8
#> [31] aplot_0.1.10 ash_1.0-15 GenomeInfoDb_1.36.0
#> [34] jsonlite_1.8.5 progress_1.2.2 highr_0.10
#> [37] tweenr_2.0.2 parallel_4.3.0 prettyunits_1.1.1
#> [40] R6_2.5.1 bslib_0.5.0 ggmsa_1.3.4
#> [43] stringi_1.7.12 extrafontdb_1.0 jquerylib_0.1.4
#> [46] Rcpp_1.0.10 knitr_1.43 iNEXT_3.0.0
#> [49] VennDiagram_1.7.3 extrafont_0.19 IRanges_2.34.0
#> [52] Matrix_1.5-4 igraph_1.4.3 timechange_0.2.0
#> [55] tidyselect_1.2.0 stringdist_0.9.10 yaml_2.3.7
#> [58] codetools_0.2-19 plyr_1.8.8 lattice_0.21-8
#> [61] treeio_1.25.0.001 withr_2.5.0 evaluate_0.21
#> [64] lambda.r_1.2.4 gridGraphics_0.5-1 desc_1.4.2
#> [67] futile.logger_1.4.3 polyclip_1.10-4 circlize_0.4.15
#> [70] Biostrings_2.68.1 pillar_1.9.0 ggtree_3.9.0
#> [73] KernSmooth_2.23-20 stats4_4.3.0 ggfun_0.0.9
#> [76] generics_0.1.3 rprojroot_2.0.3 dtplyr_1.3.1
#> [79] RCurl_1.98-1.12 S4Vectors_0.38.1 hms_1.1.3
#> [82] munsell_0.5.0 scales_1.2.1 tidytree_0.4.2
#> [85] glue_1.6.2 pheatmap_1.0.12 lazyeval_0.2.2
#> [88] tools_4.3.0 fs_1.6.2 fastmatch_1.1-3
#> [91] grid_4.3.0 ape_5.7-1 Rttf2pt1_1.3.12
#> [94] R4RNA_1.28.0 colorspace_2.1-0 nlme_3.1-162
#> [97] GenomeInfoDbData_1.2.10 patchwork_1.1.2 ggalt_0.4.0
#> [100] ggforce_0.4.1 cli_3.6.1 msa_1.32.0
#> [103] textshaping_0.3.6 futile.options_1.0.1 ineq_0.2-13
#> [106] proj4_1.0-12 fansi_1.0.4 gtable_0.3.3
#> [109] yulab.utils_0.0.6 sass_0.4.6 digest_0.6.31
#> [112] BiocGenerics_0.46.0 ggplotify_0.1.0 htmlwidgets_1.6.2
#> [115] farver_2.1.1 memoise_2.0.1 htmltools_0.5.5
#> [118] pkgdown_2.0.7 lifecycle_1.0.3 GlobalOptions_0.1.2
#> [121] bit64_4.0.5 MASS_7.3-58.4