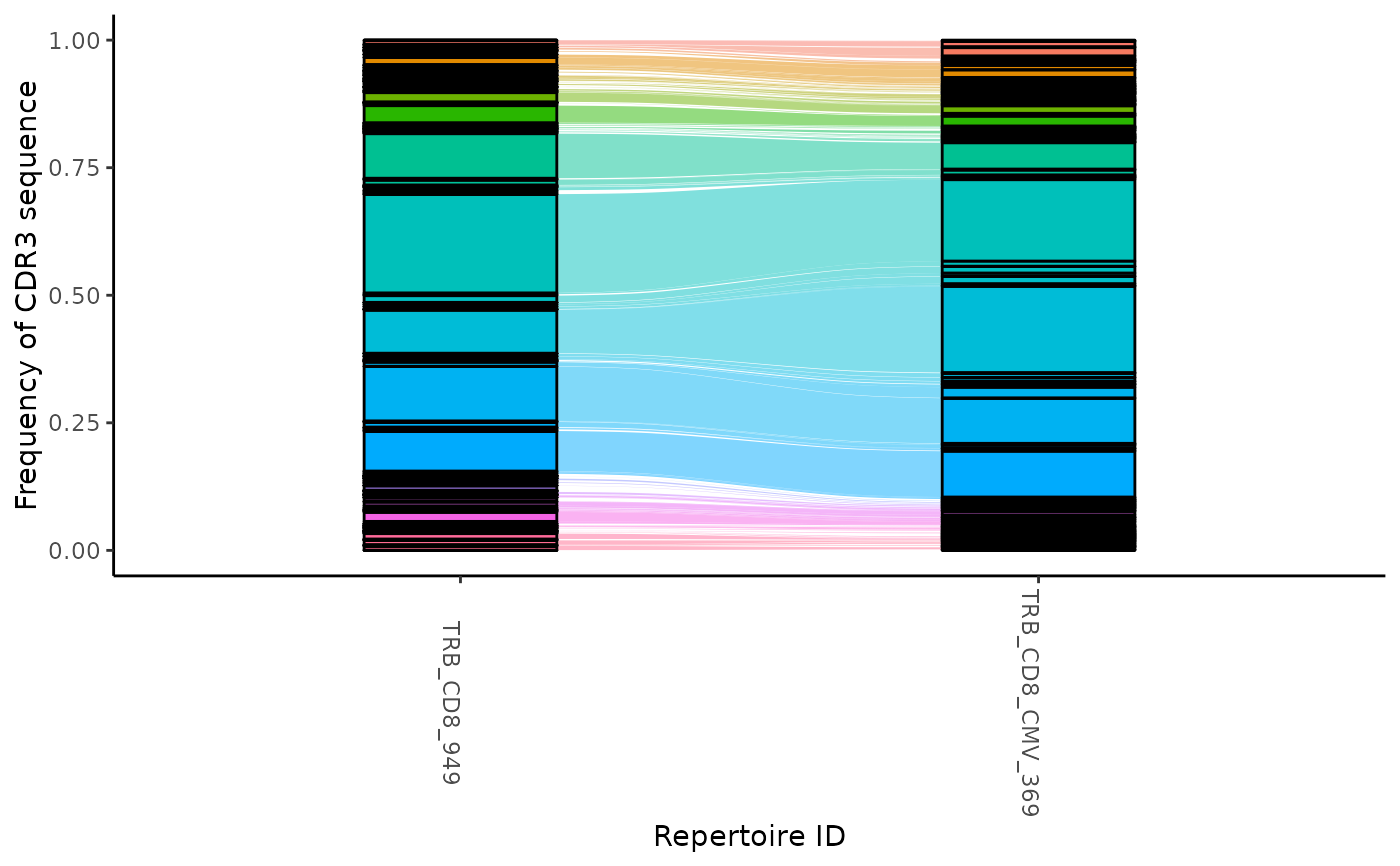
Creates alluvial tracking amino acid frequencies across multiple samples
Usage
plotTrack(
clone_table,
alist = NULL,
apal = NULL,
breaks = 2,
alphas = 0,
breaks_pal = c("#7fc97f", "#beaed4")
)Arguments
- clone_table
A tibble of productive amino acid sequences generated by LymphoSeq function cloneTrack
- alist
An optional list of amino acid, if a list is provided only those sequences will be highlighted
- apal
An optional list of palatte colors used for the amino acids to be highlighted
- breaks
Add an additional band behind the alluvial plot to highligh group, specificies after which bar the break should appear
- alphas
Set alpha for the band
- breaks_pal
Specify palette for the bands
Details
The plot is made using the package ggplot2 and can be reformatted using ggplot2 functions. See examples below.
Examples
file_path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeqTest")
stable <- LymphoSeqTest::readImmunoSeq(path = file_path)
#> Rows: 1 Columns: 144
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (69): sequence_id, sequence, sequence_aa, locus, v_call, d_call, d2_call...
#> dbl (70): v_score, v_identity, v_support, d_score, d_identity, d_support, d2...
#> lgl (5): rev_comp, productive, vj_in_frame, stop_codon, complete_vdj
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (33): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (4): vFamilyTies, jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 414 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (35): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (2): jFamilyTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 920 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
atable <- LymphoSeqTest::productiveSeq(stable, aggregate = "junction_aa")
ctable <- LymphoSeqTest::cloneTrack(study_table = atable,
sample_list = c("TRB_CD8_949", "TRB_CD8_CMV_369"))
LymphoSeqTest::plotTrack(ctable)