Create a phylogenetic tree using neighbor joining tree estimation for amino acid or junction CDR3 sequences in a list of data frames.
Usage
phyloTree(
study_table,
repertoire_ids,
type = "junction",
layout = "rectangular",
label = TRUE
)Arguments
- study_table
A tibble of unproductive junction sequences or productive junction sequences generated by the LymphoSeq function productiveSeq. v_family, d_family, j_family, and duplicate_count are required columns.
- repertoire_ids
A character vector indicating the name of the repertoire_id in the study table.
- type
A character vector indicating whether "junction_aa" or "junction" sequences should be compared.
- layout
A character vector indicating the tree layout. Options include "rectangular", "slanted", "fan", "circular", "radial" and "unrooted".
- label
A Boolean indicating if the sequencing duplicate_count should be shown next to the leaves.
Value
Returns a phylogenetic tree where each leaf represents a sequence color coded by the V, D, and J gene usage. The number next to each leaf refers to the sequence duplicate_count. A triangle shaped leaf indicates the dominant sequence. Refer to the ggtree Bioconductor package documentation for details on how to manipulate the tree.
Examples
file_path <- system.file("extdata", "IGH_sequencing", package = "LymphoSeqTest")
stable <- readImmunoSeq(path = file_path)
#> Rows: 1 Columns: 144
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (69): sequence_id, sequence, sequence_aa, locus, v_call, d_call, d2_call...
#> dbl (70): v_score, v_identity, v_support, d_score, d_identity, d_support, d2...
#> lgl (5): rev_comp, productive, vj_in_frame, stop_codon, complete_vdj
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 694 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (10): vFamilyTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFun...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 41 rows [14, 15, 33, 36, 48, 78, 119, 123, 130, 135, 149, 167, 176, 190, 198, 210, 245, 247, 250, 262, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (8): vFamilyTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFun...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 58 rows [31, 33, 40, 41, 90, 96, 109, 117, 146, 154, 178, 189, 238, 252, 255, 260, 270, 278, 315, 320, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 694 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (10): vFamilyTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFun...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 41 rows [14, 15, 33, 36, 48, 78, 119, 123, 130, 135, 149, 167, 176, 190, 198, 210, 245, 247, 250, 262, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 694 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (26): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (9): vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFunction, fracti...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 10 rows [204, 206, 265, 347, 410, 411, 419, 512, 582, 608].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 492 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (18): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (9): jGeneAlleleTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, ...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 3 rows [134, 143, 251].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 209 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (10): jGeneAlleleTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, ...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 20 rows [4, 27, 34, 37, 52, 53, 55, 69, 81, 87, 88, 90, 95, 108, 111, 131, 151, 158, 160, 200].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 436 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (25): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (10): jGeneAlleleTies, vOrphon, dOrphon, jOrphon, vFunction, dFunction, ...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 47 rows [21, 22, 28, 59, 63, 69, 78, 79, 82, 87, 90, 91, 116, 121, 149, 170, 182, 188, 216, 237, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (26): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (17): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (9): vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFunction, fracti...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 27 rows [117, 121, 146, 157, 178, 199, 296, 310, 322, 323, 324, 325, 349, 351, 363, 420, 421, 467, 468, 484, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (26): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (18): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (8): vOrphon, dOrphon, jOrphon, vFunction, dFunction, jFunction, vAlign...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 85 rows [38, 58, 79, 83, 92, 119, 127, 145, 149, 161, 162, 169, 187, 191, 199, 237, 250, 272, 275, 283, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 275 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (24): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (18): count (reads), frequencyCount (%), cdr3Length, vDeletion, n1Insert...
#> lgl (10): vFamilyTies, jGeneAlleleTies, vOrphon, dOrphon, jOrphon, vFunction...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Warning: Expected 2 pieces. Additional pieces discarded in 24 rows [9, 29, 40, 42, 61, 84, 87, 101, 104, 106, 108, 119, 146, 170, 177, 192, 201, 206, 214, 248, ...].
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
ntable <- productiveSeq(study_table = stable, aggregate = "junction")
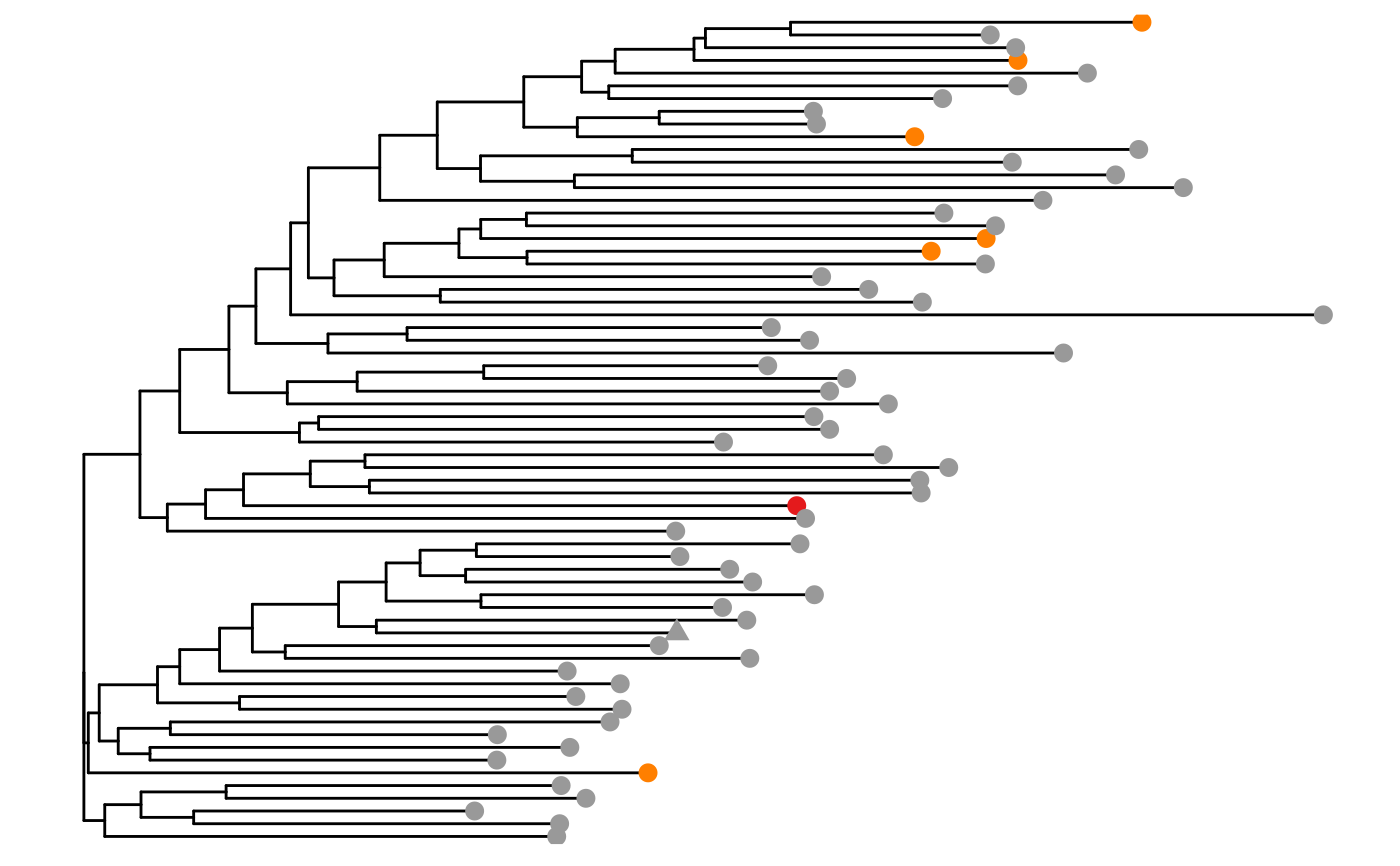
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction",
layout = "rectangular")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
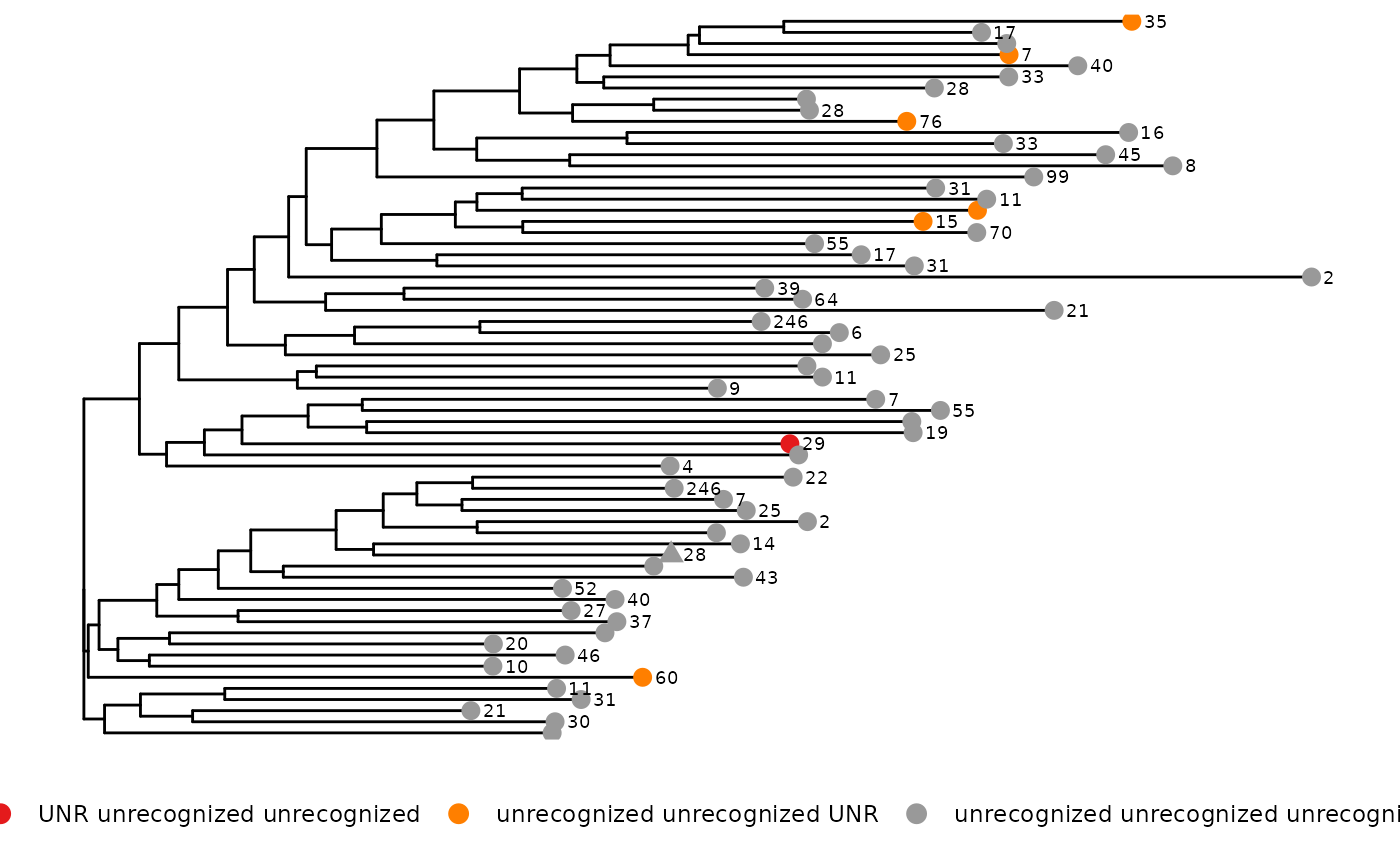 phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction_aa",
layout = "circular")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
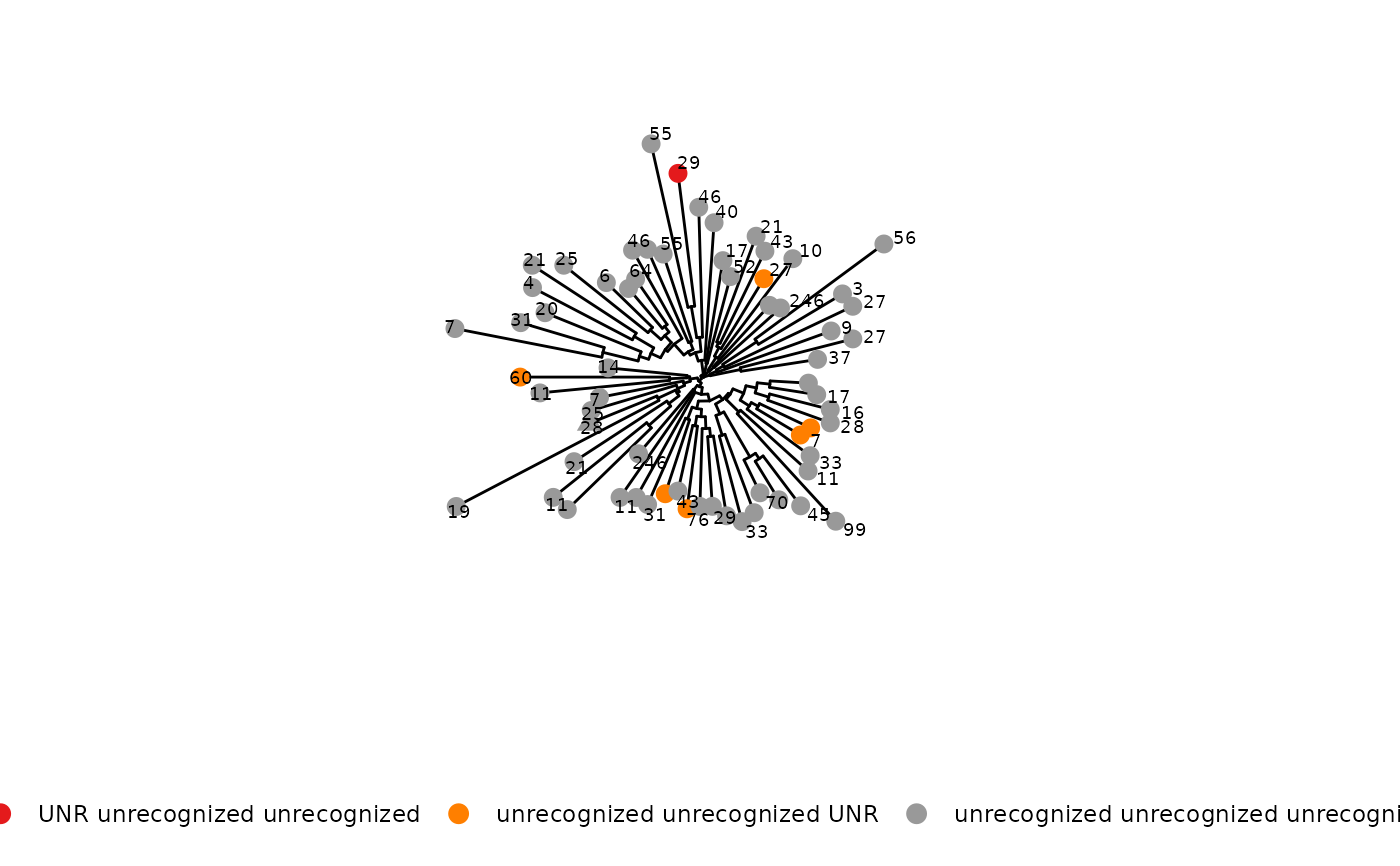
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction_aa",
layout = "circular")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
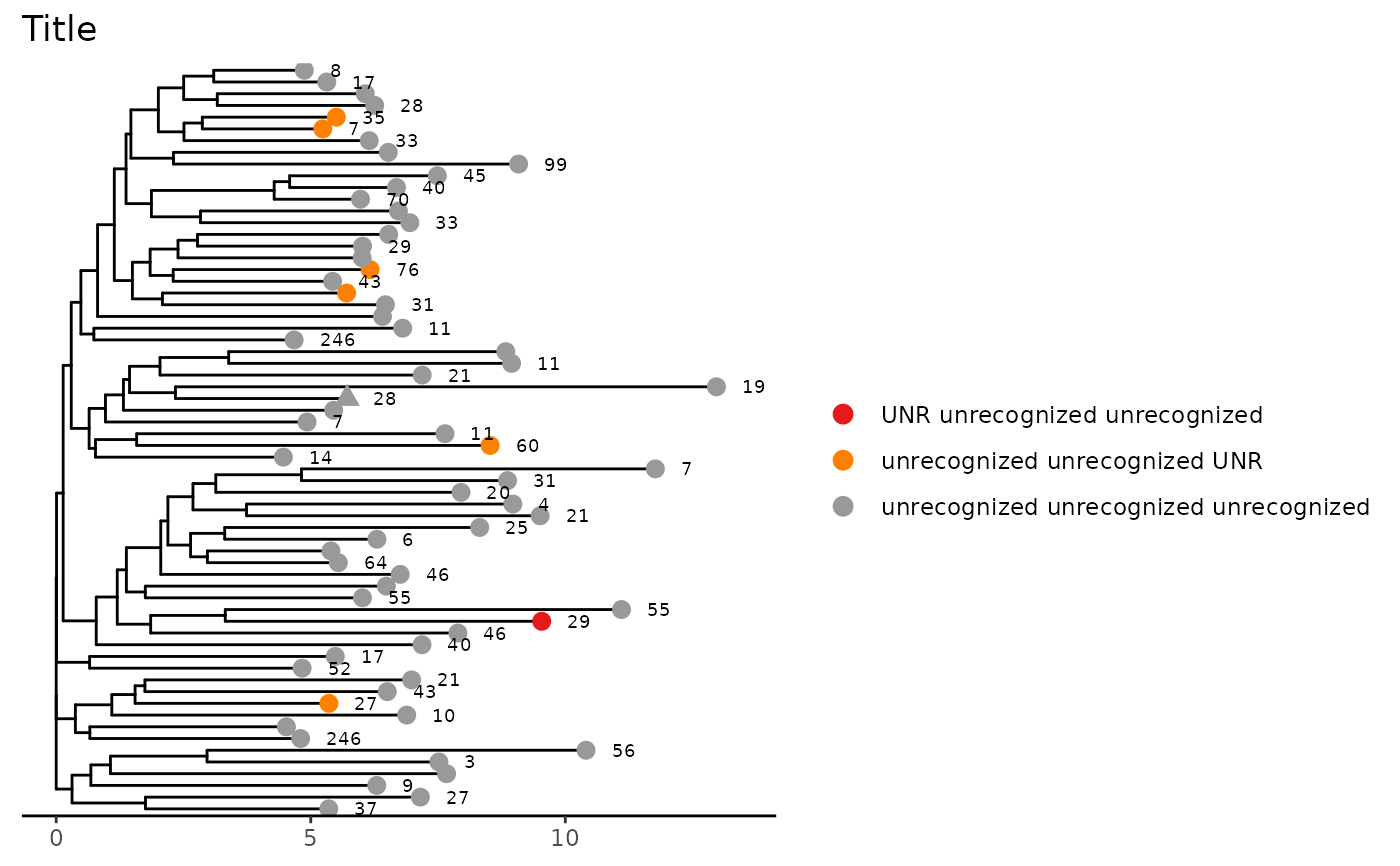
 # Add scale and title to figure
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction_aa",
layout = "rectangular") +
ggtree::theme_tree2() +
ggplot2::theme(legend.position = "right", legend.key = ggplot2::element_rect(colour = "white")) +
ggplot2::ggtitle("Title")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
# Add scale and title to figure
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction_aa",
layout = "rectangular") +
ggtree::theme_tree2() +
ggplot2::theme(legend.position = "right", legend.key = ggplot2::element_rect(colour = "white")) +
ggplot2::ggtitle("Title")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
 # Hide legend and leaf labels
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction",
layout = "rectangular", label = FALSE) +
ggplot2::theme(legend.position="none")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.
# Hide legend and leaf labels
phyloTree(study_table = ntable, repertoire_ids = "IGH_MVQ92552A_BL", type = "junction",
layout = "rectangular", label = FALSE) +
ggplot2::theme(legend.position="none")
#> Found more than one class "phylo" in cache; using the first, from namespace 'phyloseq'
#> Also defined by ‘tidytree’
#> Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> = "none")` instead.