Creates a heat map from a Bhattacharyya, Similarity, Sorensen, or PSI matrix.
Arguments
- matrix
A Bhattacharyya, Similarity, Sorensen, or PSI matrix produced by the LymphoSeq2 scoringMatrix function.
Details
The plot is made using the package ggplot2 and can be reformatted using ggplot2 functions. See examples below.
See also
An excellent resource for examples on how to reformat a ggplot can
be found in the R Graphics Cookbook online (http://www.cookbook-r.com/Graphs/).
The functions to create the similarity or Bhattacharyya matrix can be found
here: similarityMatrix and bhattacharyyaMatrix
Examples
file_path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeqTest")
stable <- readImmunoSeq(path = file_path)
#> Rows: 1 Columns: 144
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (69): sequence_id, sequence, sequence_aa, locus, v_call, d_call, d2_call...
#> dbl (70): v_score, v_identity, v_support, d_score, d_identity, d_support, d2...
#> lgl (5): rev_comp, productive, vj_in_frame, stop_codon, complete_vdj
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (33): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (4): vFamilyTies, jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 414 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (35): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (2): jFamilyTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 920 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
atable <- productiveSeq(stable, aggregate = "junction_aa")
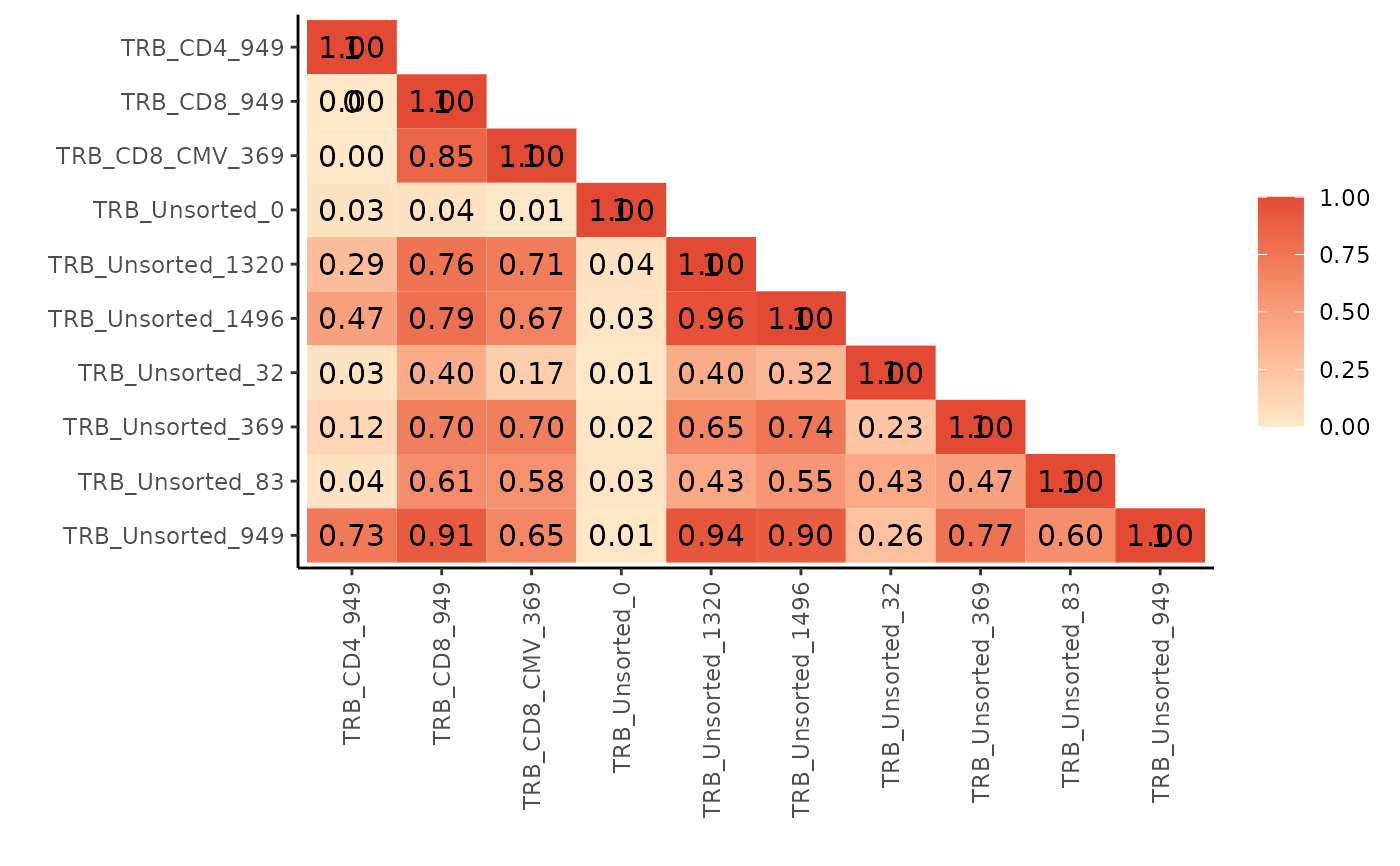
similarity_matrix <- scoringMatrix(productive_table = atable, mode="Similarity")
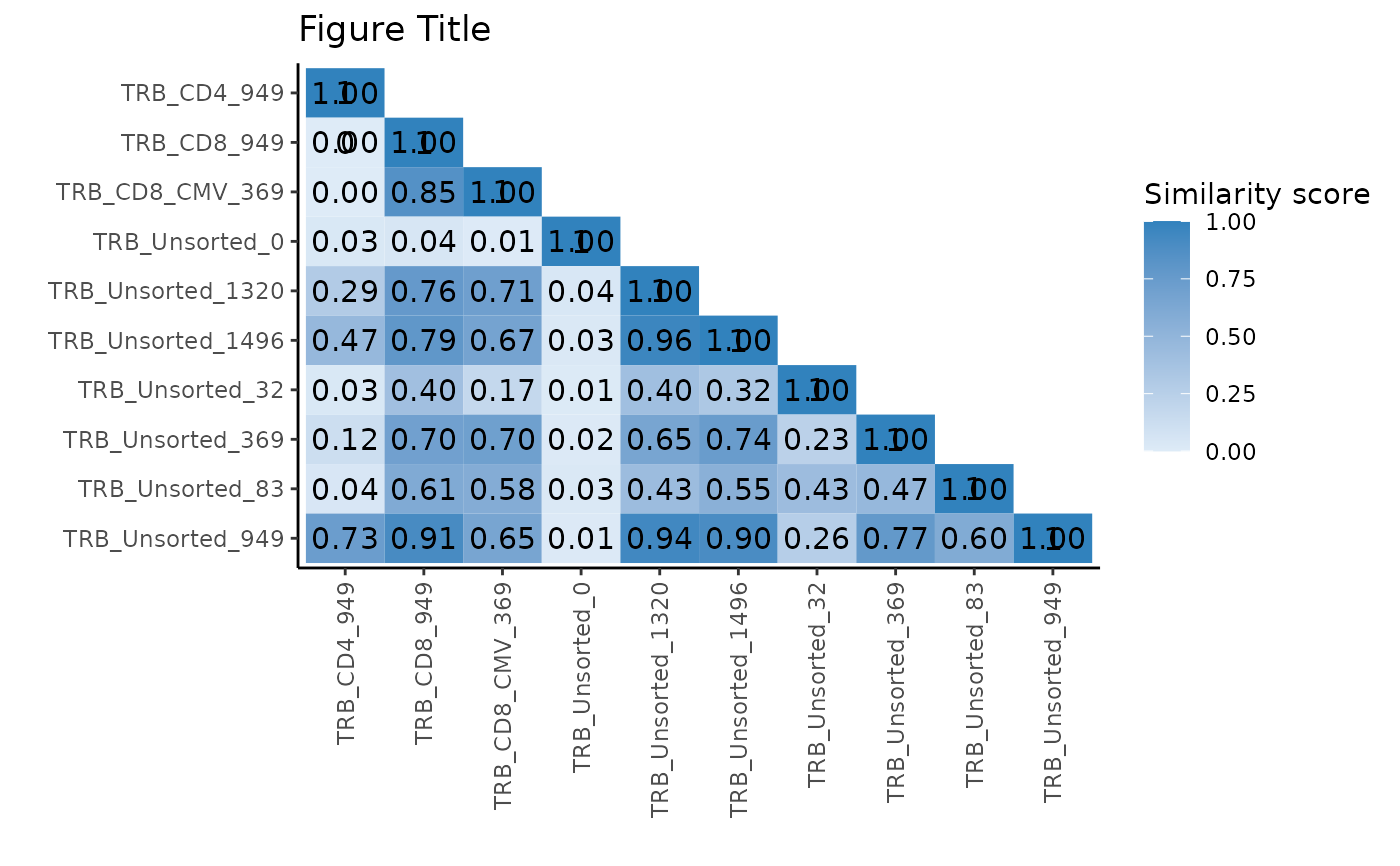
pairwisePlot(matrix = similarity_matrix)
 bhattacharyya_matrix <- scoringMatrix(productive_table = atable, mode="Bhattacharyya")
#> Error in tic(): could not find function "tic"
pairwisePlot(matrix = bhattacharyya_matrix)
#> Error in rownames(matrix): object 'bhattacharyya_matrix' not found
# Change plot color, title legend, and add title
pairwisePlot(matrix = similarity_matrix) +
ggplot2::scale_fill_gradient(low = "#deebf7", high = "#3182bd") +
ggplot2::labs(fill = "Similarity score") +
ggplot2::ggtitle("Figure Title")
#> Scale for 'fill' is already present. Adding another scale for 'fill', which
#> will replace the existing scale.
bhattacharyya_matrix <- scoringMatrix(productive_table = atable, mode="Bhattacharyya")
#> Error in tic(): could not find function "tic"
pairwisePlot(matrix = bhattacharyya_matrix)
#> Error in rownames(matrix): object 'bhattacharyya_matrix' not found
# Change plot color, title legend, and add title
pairwisePlot(matrix = similarity_matrix) +
ggplot2::scale_fill_gradient(low = "#deebf7", high = "#3182bd") +
ggplot2::labs(fill = "Similarity score") +
ggplot2::ggtitle("Figure Title")
#> Scale for 'fill' is already present. Adding another scale for 'fill', which
#> will replace the existing scale.