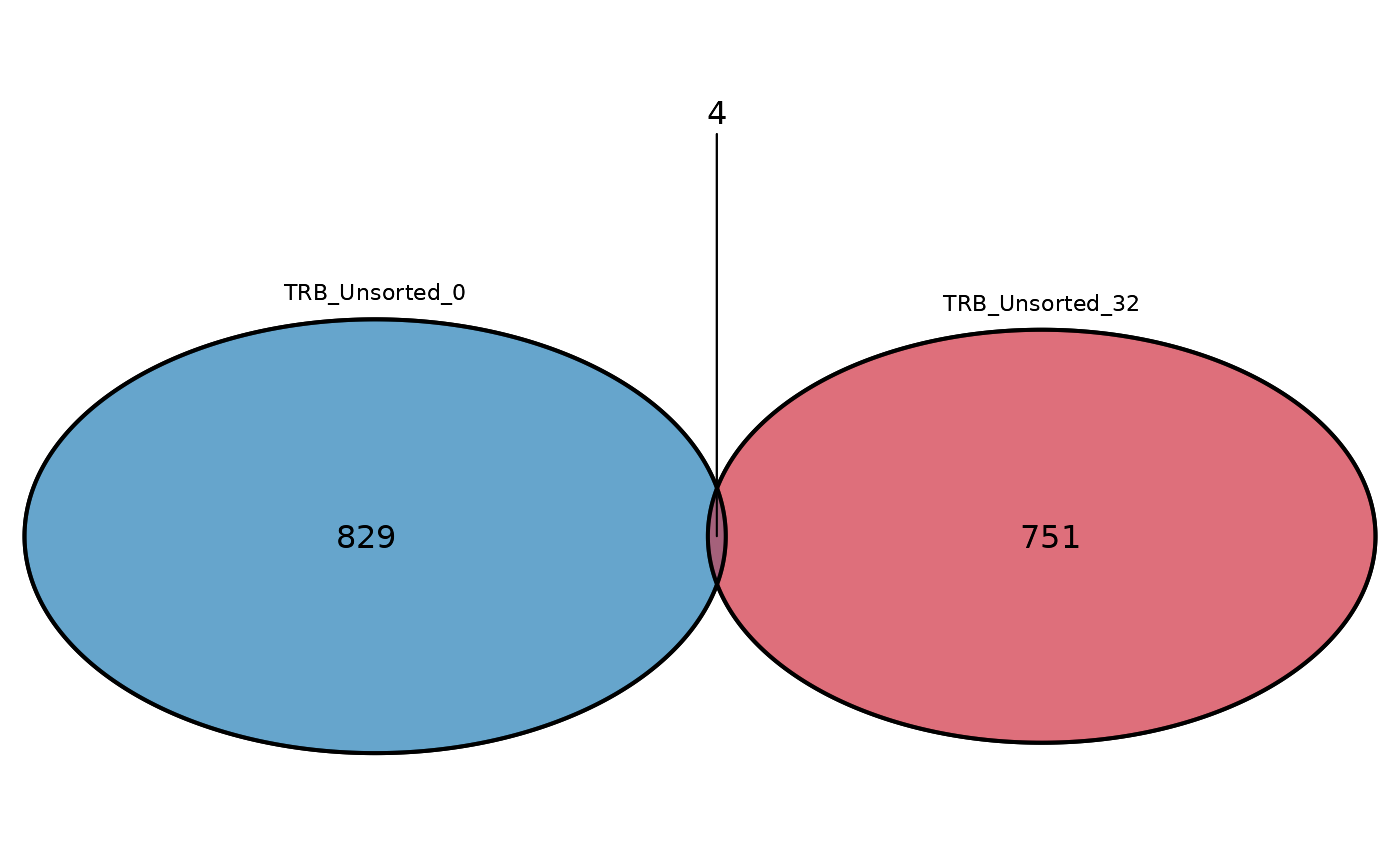
Creates a Venn diagram comparing the number of common sequences in two or three repertoire_ids.
Arguments
- repertoire_ids
A character vector of two or three names of repertoire_ids in productiveSeq table to compare.
- productive_aa
A tibble of amino acid sequences generated by the LymphoSeq function productiveSeq.
Value
Returns a a Venn diagram of the number of common sequences between two or three repertoire_ids.
Examples
file_path <- system.file("extdata", "TCRB_sequencing", package = "LymphoSeqTest")
stable <- readImmunoSeq(path = file_path)
#> Rows: 1 Columns: 144
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: ","
#> chr (69): sequence_id, sequence, sequence_aa, locus, v_call, d_call, d2_call...
#> dbl (70): v_score, v_identity, v_support, d_score, d_identity, d_support, d2...
#> lgl (5): rev_comp, productive, vj_in_frame, stop_codon, complete_vdj
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (33): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (4): vFamilyTies, jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 414 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (35): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (2): jFamilyTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 920 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (29): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vFami...
#> dbl (14): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (9): vGeneAllele, vGeneAlleleTies, dGeneAllele, dFamilyTies, dGeneAllel...
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
#> Rows: 1000 Columns: 52
#> ── Column specification ────────────────────────────────────────────────────────
#> Delimiter: "\t"
#> chr (34): nucleotide, aminoAcid, vMaxResolved, vFamilyName, vGeneName, vGene...
#> dbl (15): count (templates/reads), frequencyCount (%), cdr3Length, vDeletion...
#> lgl (3): jFamilyTies, jGeneNameTies, jGeneAlleleTies
#>
#> ℹ Use `spec()` to retrieve the full column specification for this data.
#> ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#> Joining, by = c("sequence", "sequence_aa", "v_call", "d_call", "d2_call",
#> "j_call", "junction", "junction_aa", "duplicate_count", "clone_id",
#> "repertoire_id")
atable <- productiveSeq(study_table = stable, aggregate = "junction_aa")
# Plot a triple Venn diagram
commonSeqsVenn(repertoire_ids = c("TRB_Unsorted_0",
"TRB_Unsorted_32", "TRB_Unsorted_83"),
productive_aa = atable)
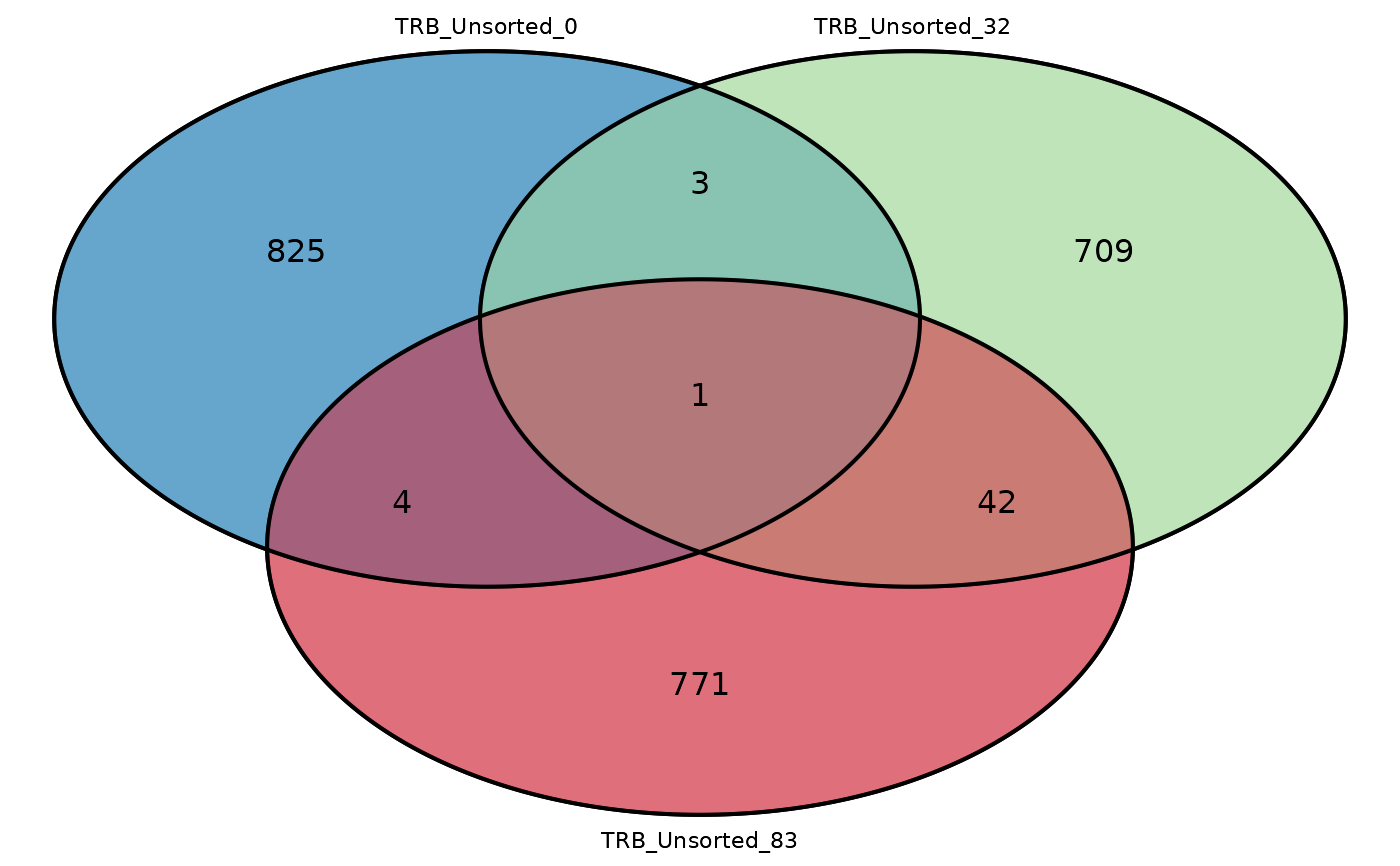 # Plot a double Venn diagram
commonSeqsVenn(repertoire_ids = c("TRB_Unsorted_0",
"TRB_Unsorted_32"), productive_aa = atable)
# Plot a double Venn diagram
commonSeqsVenn(repertoire_ids = c("TRB_Unsorted_0",
"TRB_Unsorted_32"), productive_aa = atable)